for (pkg in c("desplot", "emmeans", "ggtext", "here", "MetBrewer",
"multcomp", "multcompView", "tidyverse")) {
if (!require(pkg, character.only = TRUE)) install.packages(pkg)
}
library(desplot)
library(emmeans)
library(ggtext)
library(here)
library(MetBrewer)
library(multcomp)
library(multcompView)
library(tidyverse)Um alle in diesem Kapitel verwendeten Pakete zu installieren und zu laden, führe folgenden Code aus:
Von einem zu zwei Behandlungsfaktoren
In den Kapiteln “Linear Models in Experiments” drehte sich jede Analyse um einen einzelnen Behandlungsfaktor: eine Reihe von Sorten, Varietäten oder Genotypen, deren Mittelwerte wir vergleichen wollten. Reale Versuche variieren jedoch oft zwei Behandlungsfaktoren gleichzeitig. Ein klassisches Beispiel ist ein Düngeversuch, in dem mehrere Genotypen jeweils bei mehreren Stickstoffstufen angebaut werden. Dann interessiert uns nicht nur “welcher Genotyp ist der beste?” und “welche Stickstoffstufe ist die beste?”, sondern auch, wie die beiden Faktoren zusammenwirken.
Was ist eine Interaktion?
Mit zwei Behandlungsfaktoren stellt sich eine neue Frage, die es mit einem einzelnen Faktor schlicht nicht gab: Wirken die Faktoren unabhängig voneinander, oder hängt der Effekt des einen Faktors von der Stufe des anderen ab? Das ist die Idee einer Interaktion.
- Wenn Stickstoff den Ertrag bei jedem Genotyp um ungefähr denselben Betrag erhöht, wirken die beiden Faktoren unabhängig (keine Interaktion). Man könnte dann einen allgemeinen Stickstoffeffekt und einen allgemeinen Genotypeffekt getrennt beschreiben.
- Wenn jedoch einige Genotypen stark auf Stickstoff reagieren, während andere kaum reagieren, dann hängt der Stickstoffeffekt vom Genotyp ab. Das ist eine Interaktion (geschrieben
N:G), und sie ändert, wie wir die Ergebnisse interpretieren.
Diese Interaktion zu erkennen und zu berücksichtigen ist der ganze Sinn einer zweifaktoriellen Analyse. Wie wir sehen werden, sagt uns eine signifikante Interaktion, dass wir die einzelnen Faktorstufen-Kombinationen vergleichen sollten statt der Haupteffekte für sich allein.
Das Design bleibt ein RCBD
Die Versuchsanlage selbst ist nichts Neues: Der Versuch ist als randomisierte vollständige Blockanlage (RCBD) angelegt, genau wie im Kapitel zum einfaktoriellen RCBD. Es gibt drei vollständige Blöcke (rep), und jede der 24 Stickstoff-Genotyp-Kombinationen erscheint genau einmal in jedem Block. Das Einzige, was sich gegenüber dem früheren RCBD-Kapitel ändert, ist, dass unsere Behandlung nun eine faktorielle Kombination zweier Faktoren statt eines einzelnen ist.
Daten
Dieser Datensatz ist eine leicht modifizierte Version eines Ertragsversuchs (kg/ha), der ursprünglich in Gomez und Gomez (1984) veröffentlicht wurde. Er kreuzt 4 Genotypen (G) mit 6 Stickstoffstufen (N), was 24 Behandlungskombinationen ergibt, jede wiederholt in 3 vollständigen Blöcken (rep) - insgesamt 72 Parzellen.
Import
Rows: 72 Columns: 6
── Column specification ────────────────────────────────────────────────────────
Delimiter: ","
chr (3): rep, N, G
dbl (3): row, col, yield
ℹ Use `spec()` to retrieve the full column specification for this data.
ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.# A tibble: 72 × 6
row col rep N G yield
<dbl> <dbl> <chr> <chr> <chr> <dbl>
1 2 6 rep1 Goomba Simba 4520
2 3 4 rep1 Koopa Simba 5598
3 2 3 rep1 Toad Simba 6192
4 1 1 rep1 Peach Simba 8542
5 2 1 rep1 Diddy Simba 5806
6 3 1 rep1 Yoshi Simba 7470
7 4 5 rep1 Goomba Nala 4034
8 4 1 rep1 Koopa Nala 6682
9 3 2 rep1 Toad Nala 6869
10 1 2 rep1 Peach Nala 6318
# ℹ 62 more rowsDer Datensatz enthält:
-
N: Sechs Stickstoffstufen (der erste Behandlungsfaktor) -
G: Vier Genotypen (der zweite Behandlungsfaktor) -
rep: Drei vollständige Blöcke -
yield: Ernteertrag in kg/ha -
rowundcol: Feldparzellenkoordinaten für die Visualisierung mit desplot
Format
Wie immer sollten die kategorialen Spalten als Faktoren codiert werden. Hier betrifft das den Block (rep) und beide Behandlungsfaktoren (N und G):
# A tibble: 72 × 6
row col rep N G yield
<dbl> <dbl> <fct> <fct> <fct> <dbl>
1 2 6 rep1 Goomba Simba 4520
2 3 4 rep1 Koopa Simba 5598
3 2 3 rep1 Toad Simba 6192
4 1 1 rep1 Peach Simba 8542
5 2 1 rep1 Diddy Simba 5806
6 3 1 rep1 Yoshi Simba 7470
7 4 5 rep1 Goomba Nala 4034
8 4 1 rep1 Koopa Nala 6682
9 3 2 rep1 Toad Nala 6869
10 1 2 rep1 Peach Nala 6318
# ℹ 62 more rowsErkunden
Fassen wir zunächst den Ertrag getrennt für jeden Behandlungsfaktor zusammen. Das gibt einen ersten Eindruck von den beiden Haupteffekten:
# A tibble: 6 × 4
N count mean_yield sd_yield
<fct> <int> <dbl> <dbl>
1 Diddy 12 5866. 832.
2 Toad 12 5864. 1434.
3 Yoshi 12 5812 2349.
4 Peach 12 5797. 2660.
5 Koopa 12 5478. 657.
6 Goomba 12 4054. 672.# A tibble: 4 × 4
G count mean_yield sd_yield
<fct> <int> <dbl> <dbl>
1 Simba 18 6554. 1475.
2 Nala 18 6156. 1078.
3 Timon 18 5563. 1269.
4 Pumba 18 3642. 1434.Diese einfaktoriellen Zusammenfassungen sind nützlich, verbergen aber genau das, was uns am meisten interessiert: ob der Stickstoffeffekt für jeden Genotyp derselbe ist. Um das zu sehen, müssen wir die Faktorstufen-Kombinationen betrachten. Eine bequeme Möglichkeit dazu ist, den Ertrag gegen Stickstoff aufzutragen und ein Panel (Facette) je Genotyp zu zeichnen.
Zunächst definieren wir einen festen Satz Farben für die sechs Stickstoffstufen, den wir im gesamten Kapitel wiederverwenden:
ggplot(data = dat) +
aes(y = yield, x = N, color = N) +
facet_wrap(~G, labeller = label_both) +
stat_summary(
fun = mean,
colour = "grey",
geom = "line",
linetype = "dotted",
group = 1
) +
geom_point() +
scale_x_discrete(name = "Stickstoff") +
scale_y_continuous(
name = "Ertrag",
limits = c(0, NA),
expand = expansion(mult = c(0, 0.1))
) +
scale_color_manual(values = Ncolors, guide = "none") +
theme_bw() +
theme(axis.text.x = element_text(angle = 45, hjust = 1, vjust = 1))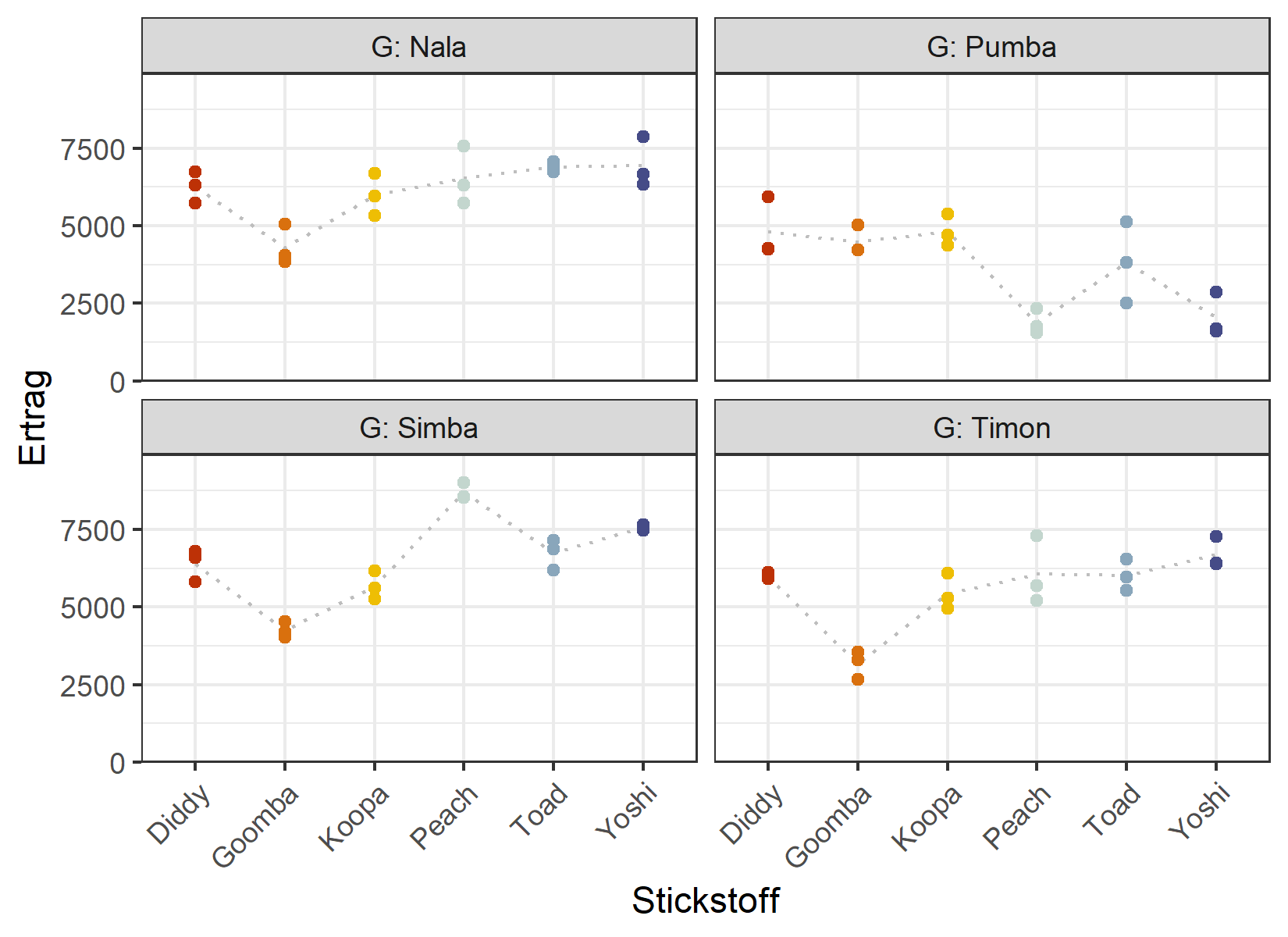
Die gepunktete graue Linie verbindet den mittleren Ertrag je Stickstoffstufe innerhalb jedes Genotyps. Würden die Faktoren perfekt unabhängig wirken, hätten diese Profile in jedem Panel dieselbe Form. Das tun sie offensichtlich nicht - Rangfolge und Abstände der Stickstoffstufen unterscheiden sich zwischen den Genotypen. Das ist ein erster visueller Hinweis auf eine Interaktion, die wir in der ANOVA formal testen werden.
Da es sich um einen geplanten Versuch handelt, lohnt sich auch ein Blick auf das Feld-Layout. Das geht mit desplot() aus dem Paket {desplot}. Hier färben wir die Genotyp-Beschriftungen nach ihrer Stickstoffstufe und ziehen Linien zwischen den Blöcken:
desplot(
data = dat,
form = rep ~ col + row | rep, # ein Panel je Block
col.regions = c("white", "grey95", "grey90"),
text = G, # Genotypnamen je Parzelle
cex = 0.8, # Genotypnamen: Schriftgröße
shorten = "abb", # Genotypnamen: abkürzen
col = N, # Genotypnamen nach Stickstoffstufe färben
col.text = Ncolors, # die eigenen Stickstofffarben verwenden
out1 = col, out1.gpar = list(col = "darkgrey"), # Linien zwischen Spalten
out2 = row, out2.gpar = list(col = "darkgrey"), # Linien zwischen Reihen
main = "Feld-Layout",
show.key = TRUE,
key.cex = 0.7
)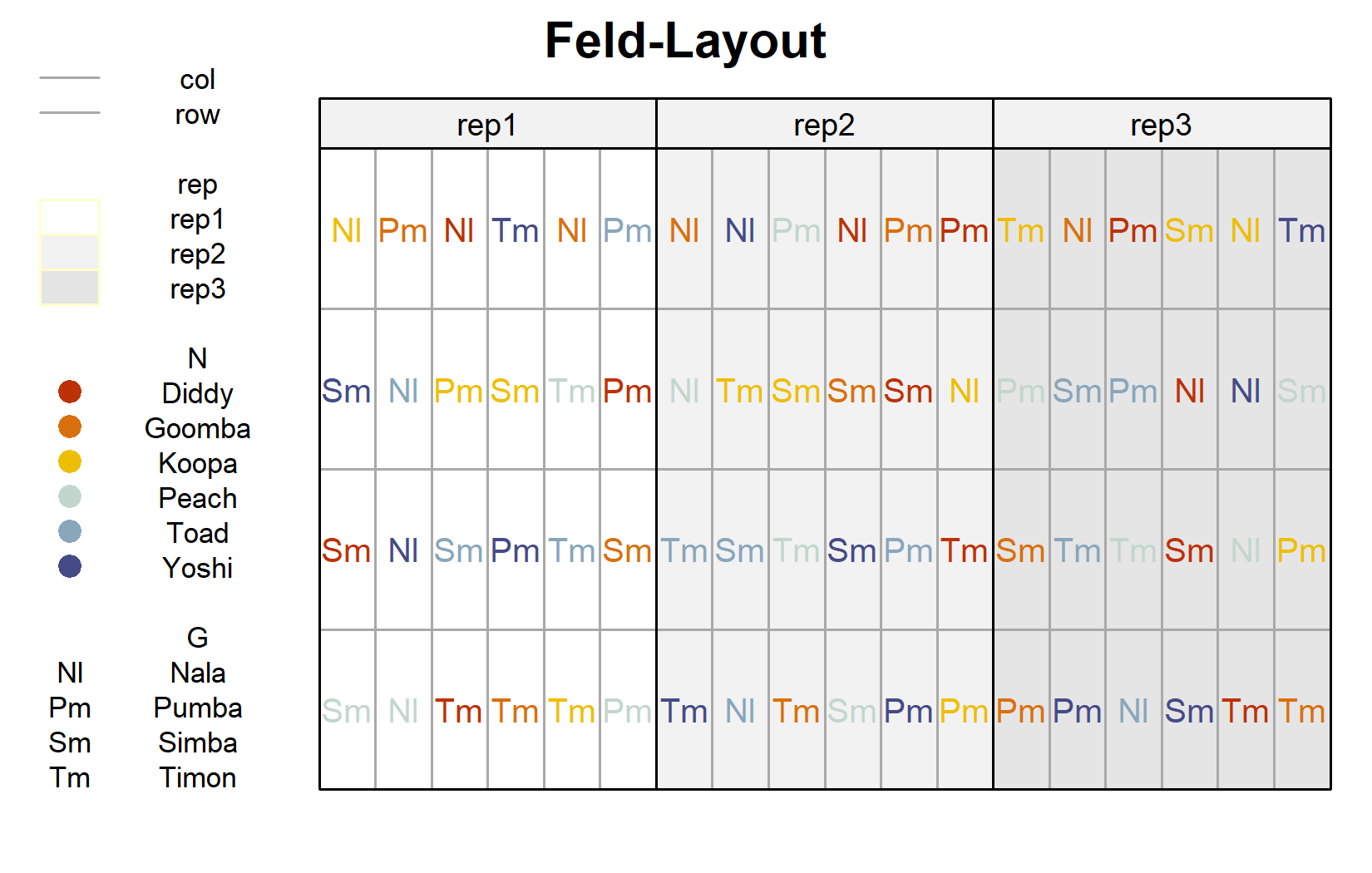
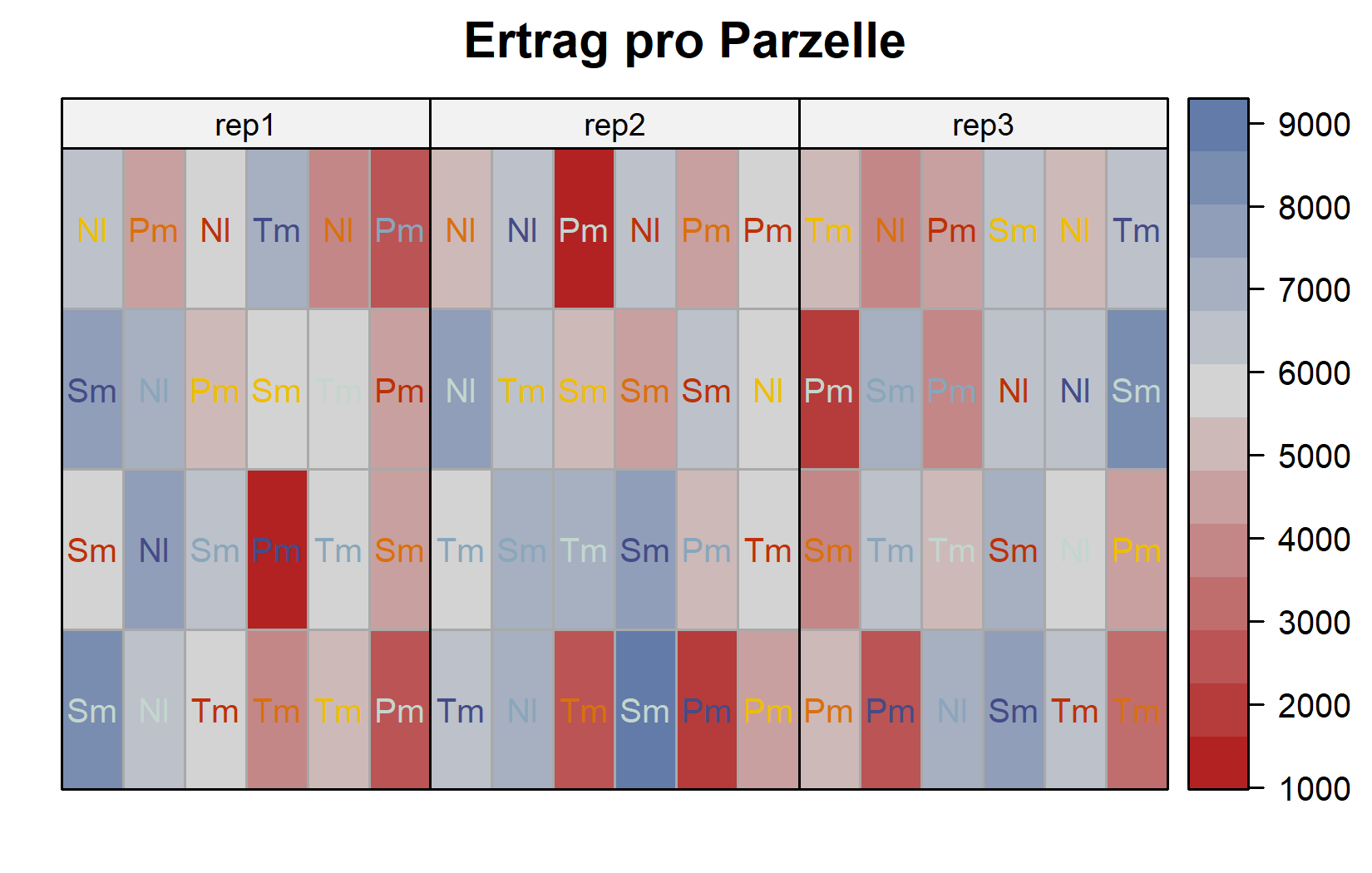
Das Layout bestätigt, dass jede Stickstoff-Genotyp-Kombination einmal je Block erscheint und dass die 24 Kombinationen innerhalb jedes der drei Blöcke frisch randomisiert sind - das definierende Merkmal eines RCBD.
Modell
Wir fitten nun ein lineares Modell mit dem Ertrag als Zielgröße. Es hilft, die Modellterme gedanklich in Behandlungseffekte und Designeffekte zu gruppieren. Die Behandlungen sind hier die Genotypen G und die Stickstoffstufen N, die wir als Haupteffekte und über ihre Interaktion N:G einbeziehen. Das Design steuert den Blockeffekt rep bei.
mod <- lm(yield ~ N + G + N:G + rep, data = dat)Verglichen mit dem einfaktoriellen RCBD-Modell yield ~ cultivar + block ist die einzige strukturelle Ergänzung der zweite Behandlungsfaktor und der Interaktionsterm N:G. Alles andere - das Fitten mit lm(), das Durchführen von anova(), das Berechnen adjustierter Mittel mit emmeans() - funktioniert genau wie zuvor.
WarnungModellannahmen erfüllt?
An dieser Stelle (d.h. nach dem Modell-Fit und vor der ANOVA-Interpretation) sollte man prüfen, ob die Modellannahmen erfüllt sind. Mehr dazu im Anhang A1: Modelldiagnostik.
ANOVA
Auf Basis unseres Modells können wir eine ANOVA durchführen:
ANOVA <- anova(mod)
ANOVAAnalysis of Variance Table
Response: yield
Df Sum Sq Mean Sq F value Pr(>F)
N 5 30480453 6096091 15.4677 6.509e-09 ***
G 3 89885035 29961678 76.0221 < 2.2e-16 ***
rep 2 1084820 542410 1.3763 0.2627
N:G 15 69378044 4625203 11.7356 4.472e-11 ***
Residuals 46 18129432 394118
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1Die ANOVA-Tabelle enthält nun eine Zeile für jeden Modellterm, einschließlich der Interaktion N:G. In diesem Versuch ist die Interaktion statistisch signifikant (siehe ihren p-Wert in der Tabelle oben), was den Eindruck aus unserer Erkundungsgrafik bestätigt: Der Effekt von Stickstoff ist nicht für jeden Genotyp derselbe.
Wichtig
Eine signifikante Interaktion hat eine direkte Konsequenz für die Interpretation. Wenn N:G signifikant ist, sollten wir die Haupteffekte N und G nicht für sich allein interpretieren, weil der Effekt des einen Faktors von der Stufe des anderen abhängt. Stattdessen sollte sich der Mittelwertvergleich auf die einzelnen N:G-Kombinationen konzentrieren.
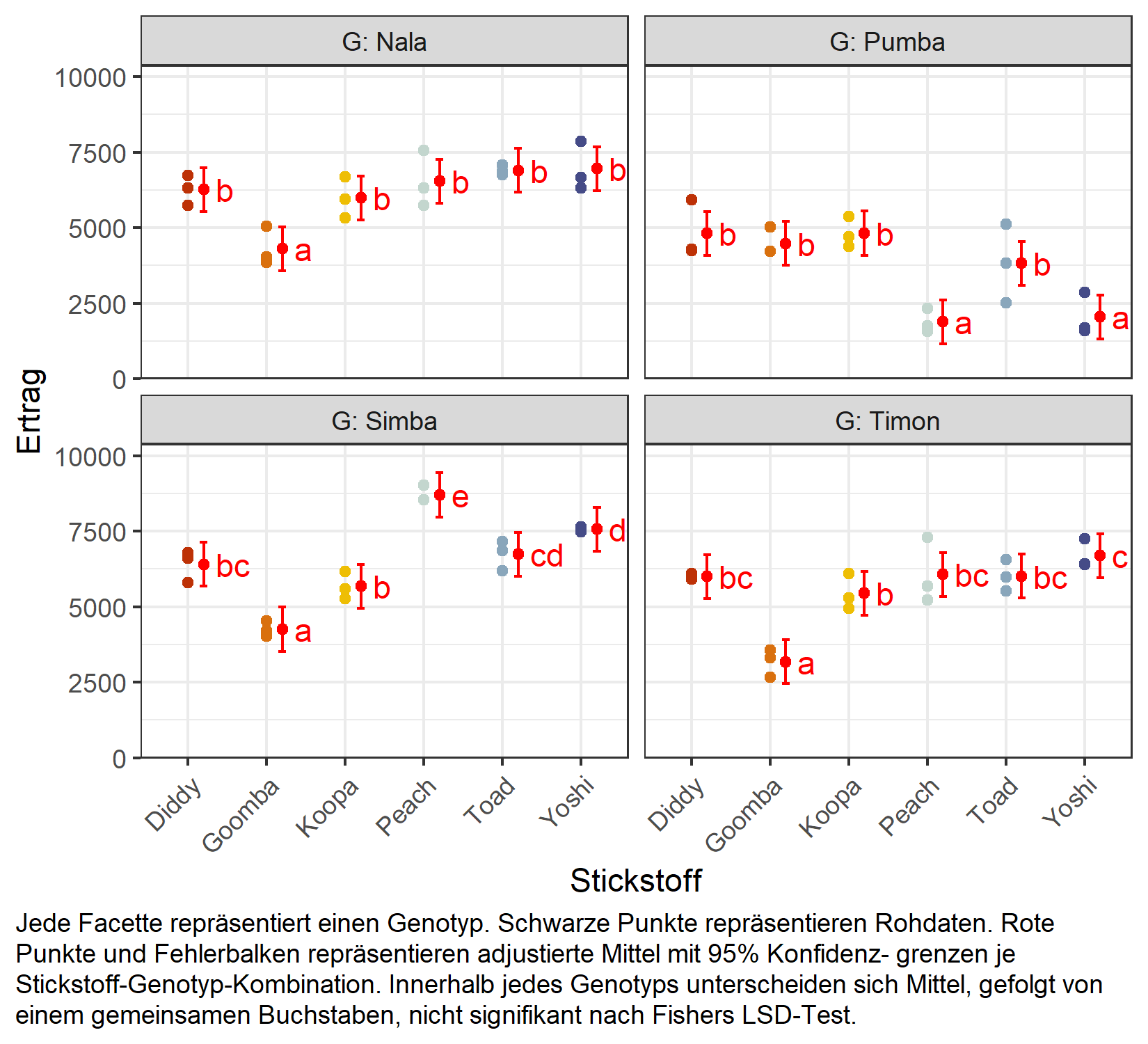
Mittelwertvergleich
Wegen der signifikanten Interaktion vergleichen wir adjustierte Mittel für die Faktorstufen-Kombinationen statt für die Haupteffekte. Eine natürliche und gut lesbare Möglichkeit dazu ist, die Stickstoffstufen innerhalb jedes Genotyps zu vergleichen, was specs = ~ N | G anfordert:
G = Nala:
N emmean SE df lower.CL upper.CL .group
Goomba 4306 362 46 3576 5036 a
Koopa 5982 362 46 5252 6712 b
Diddy 6259 362 46 5529 6989 b
Peach 6540 362 46 5811 7270 b
Toad 6895 362 46 6165 7625 b
Yoshi 6951 362 46 6221 7680 b
G = Pumba:
N emmean SE df lower.CL upper.CL .group
Peach 1881 362 46 1151 2610 a
Yoshi 2047 362 46 1317 2776 a
Toad 3816 362 46 3086 4546 b
Goomba 4481 362 46 3752 5211 b
Diddy 4812 362 46 4082 5542 b
Koopa 4816 362 46 4086 5546 b
G = Simba:
N emmean SE df lower.CL upper.CL .group
Goomba 4253 362 46 3523 4982 a
Koopa 5672 362 46 4942 6402 b
Diddy 6400 362 46 5670 7130 bc
Toad 6733 362 46 6003 7462 cd
Yoshi 7563 362 46 6834 8293 d
Peach 8701 362 46 7971 9430 e
G = Timon:
N emmean SE df lower.CL upper.CL .group
Goomba 3177 362 46 2448 3907 a
Koopa 5443 362 46 4713 6172 b
Diddy 5994 362 46 5264 6724 bc
Toad 6014 362 46 5284 6744 bc
Peach 6065 362 46 5336 6795 bc
Yoshi 6687 362 46 5958 7417 c
Results are averaged over the levels of: rep
Confidence level used: 0.95
significance level used: alpha = 0.05
NOTE: If two or more means share the same grouping symbol,
then we cannot show them to be different.
But we also did not show them to be the same. Hier lassen wir die Vergleiche unadjustiert (adjust = "none", d.h. Fishers LSD); für einen Überblick, wann und wie man für Multiplizität korrigiert, siehe Anhang A4: Multiplizitätskorrekturen. Das Ergebnis wird als kompakte Buchstabendarstellung präsentiert (siehe Anhang A5: Compact Letter Display): Innerhalb jedes Genotyps unterscheiden sich Stickstoffstufen, die einen Buchstaben teilen, nicht signifikant. Um die zugrunde liegenden paarweisen Differenzen zu sehen, fügt man einfach details = TRUE zum cld()-Aufruf hinzu.
Abschließend erstellen wir eine Grafik, die sowohl die Rohdaten als auch die Ergebnisse zeigt, d.h. die adjustierten Mittel mit ihren Konfidenzgrenzen und die kompakte Buchstabendarstellung, ein Panel je Genotyp:
my_caption <- "Jede Facette repräsentiert einen Genotyp. Schwarze Punkte repräsentieren
Rohdaten. Rote Punkte und Fehlerbalken repräsentieren adjustierte Mittel mit 95% Konfidenz-
grenzen je Stickstoff-Genotyp-Kombination. Innerhalb jedes Genotyps unterscheiden sich Mittel,
gefolgt von einem gemeinsamen Buchstaben, nicht signifikant nach Fishers LSD-Test."
ggplot() +
facet_wrap(~G, labeller = label_both) +
aes(x = N) +
# schwarze Punkte für die Rohdaten
geom_point(
data = dat,
aes(y = yield, color = N)
) +
# rote Punkte für die adjustierten Mittel
geom_point(
data = mean_comp,
aes(y = emmean),
color = "red",
position = position_nudge(x = 0.2)
) +
# rote Fehlerbalken für die Konfidenzgrenzen der adjustierten Mittel
geom_errorbar(
data = mean_comp,
aes(ymin = lower.CL, ymax = upper.CL),
color = "red",
width = 0.1,
position = position_nudge(x = 0.2)
) +
# rote Buchstaben
geom_text(
data = mean_comp,
aes(y = emmean, label = str_trim(.group)),
color = "red",
position = position_nudge(x = 0.35),
hjust = 0
) +
scale_x_discrete(name = "Stickstoff") +
scale_y_continuous(
name = "Ertrag",
limits = c(0, NA),
expand = expansion(mult = c(0, 0.1))
) +
scale_color_manual(values = Ncolors, guide = "none") +
theme_bw() +
labs(caption = my_caption) +
theme(
plot.caption = element_textbox_simple(margin = margin(t = 5)),
plot.caption.position = "plot",
axis.text.x = element_text(angle = 45, hjust = 1, vjust = 1)
)
Abschluss
Wir haben nun einen zweifaktoriellen Versuch in einer randomisierten vollständigen Blockanlage analysiert. Die Mechanik ist eine kleine Erweiterung des einfaktoriellen RCBD: den zweiten Behandlungsfaktor und seine Interaktion hinzufügen und dann die Interaktion bestimmen lassen, wie man die Mittel vergleicht.
HinweisZusammenfassung
Zwei Behandlungsfaktoren werden als Haupteffekte plus ein Interaktionsterm einbezogen:
yield ~ N + G + N:G + rep.Die Interaktion
N:Gfragt, ob der Effekt des einen Faktors von der Stufe des anderen abhängt. Sie ist die zentrale neue Frage in einer zweifaktoriellen Analyse.Eine signifikante Interaktion bedeutet, dass die Haupteffekte nicht für sich allein interpretiert werden sollten; man vergleicht stattdessen die Faktorstufen-Kombinationen.
emmeans(specs = ~ N | G)vergleicht Stickstoffstufen innerhalb jedes Genotyps - eine gut lesbare Möglichkeit, eine signifikante Interaktion aufzuschlüsseln.Das Design ist unverändert: Dies ist weiterhin ein RCBD, analysiert mit
lm(), nur mit einer faktoriellen Behandlungsstruktur.
In diesem Kapitel wurden alle 24 Behandlungskombinationen innerhalb jedes Blocks frisch randomisiert. Aber was, wenn einer der Faktoren auf größere Einheiten angewendet werden muss - zum Beispiel, wenn Stickstoff nur auf größeren Feldstreifen verwaltet werden kann? Dann kämen dieselben Daten aus einem anderen Design. Im nächsten Kapitel analysieren wir genau diesen Versuch erneut als Split-Plot-Design und sehen, wie das Modell und Inferenz verändert.
Literatur
Gomez, Kwanchai A, und Arturo A Gomez. 1984. Statistical procedures for agricultural research. 2. Aufl. An International Rice Research Institute book. Nashville, TN: John Wiley & Sons.
Zitat
Mit BibTeX zitieren:
@online{schmidt2026,
author = {{Dr. Paul Schmidt}},
publisher = {BioMath GmbH},
title = {1. Zweifaktorielle ANOVA in einem RCBD},
date = {2026-06-08},
url = {https://biomathcontent.netlify.app/de/content/lin_mod_adv/01_twoway_rcbd.html},
langid = {de}
}
Bitte zitieren Sie diese Arbeit als:
Dr. Paul Schmidt. 2026. “1. Zweifaktorielle ANOVA in einem
RCBD.” BioMath GmbH. June 8, 2026. https://biomathcontent.netlify.app/de/content/lin_mod_adv/01_twoway_rcbd.html.