for (pkg in c("desplot", "emmeans", "ggtext", "here", "lme4", "lmerTest",
"MetBrewer", "multcomp", "multcompView", "tidyverse")) {
if (!require(pkg, character.only = TRUE)) install.packages(pkg)
}
library(desplot)
library(emmeans)
library(ggtext)
library(here)
library(lme4)
library(lmerTest)
library(MetBrewer)
library(multcomp)
library(multcompView)
library(tidyverse)Um alle in diesem Kapitel verwendeten Pakete zu installieren und zu laden, führe folgenden Code aus:
Von einem RCBD zu einem Split-Plot
Im vorherigen Kapitel haben wir einen zweifaktoriellen Versuch analysiert - 4 Genotypen gekreuzt mit 6 Stickstoffstufen - angelegt als randomisierte vollständige Blockanlage. Dort wurden alle 24 Behandlungskombinationen innerhalb jedes Blocks frisch randomisiert, sodass jede Parzelle eine unabhängige Versuchseinheit war.
In der Praxis ist diese vollständige Randomisierung nicht immer möglich. Manche Behandlungsfaktoren lassen sich schlicht leichter auf große Einheiten als auf kleine anwenden. Stickstoffdüngung, Bewässerung, Bodenbearbeitung oder Aussaatzeitpunkt sind typische Beispiele: Sie parzellenweise zu verwalten ist unpraktisch, also werden sie auf größeren Streifen angewendet, und ein zweiter Faktor wird dann innerhalb dieser Streifen variiert. Genau für diese Situation ist ein Split-Plot-Design gemacht - und es ist das Design hinter den Daten dieses Kapitels.
Was ist ein Split-Plot-Design?
Ein Split-Plot-Design hat zwei Ebenen von Versuchseinheiten und damit zwei Randomisierungen:
- Großparzellen (whole plots): große Einheiten, denen die Stufen eines Faktors (des Großparzellen-Faktors) zufällig zugewiesen werden. In unserem Versuch werden die sechs Stickstoffstufen auf Großparzellen angewendet.
- Unterparzellen (subplots): jede Großparzelle wird in kleinere Einheiten unterteilt, denen die Stufen des zweiten Faktors (des Unterparzellen-Faktors) zufällig zugewiesen werden. Hier werden die vier Genotypen innerhalb jeder Großparzelle randomisiert.
Konkret hat unser Versuch 3 Blöcke, und innerhalb jedes Blocks liegen die 6 Stickstoffstufen auf 6 Großparzellen (insgesamt 18 Großparzellen). Jede Großparzelle wird dann in 4 Unterparzellen aufgeteilt, eine je Genotyp - 72 Parzellen, dieselben 72 Ertragswerte wie im vorherigen Kapitel, aber gemäß einer anderen Randomisierung angeordnet.
Warum ein gemischtes Modell?
Die beiden Randomisierungen erzeugen zwei Quellen zufälliger Variation: eine zwischen Großparzellen und eine zwischen Unterparzellen innerhalb einer Großparzelle. Um die Daten korrekt zu analysieren, muss das Modell dies widerspiegeln, indem es einen Zufallseffekt für die Großparzellen enthält. Das ist unser erstes praktisches gemischtes Modell: Wir fitten es mit lmer() aus dem Paket {lmerTest} und geben den Großparzellen-Zufallseffekt als (1 | rep:mainplot) an.
TippHintergrundlektüre
Für den theoretischen Hintergrund zu gemischten Modellen - warum wir manche Effekte als zufällig behandeln, was BLUEs und BLUPs sind und wie Freiheitsgrade approximiert werden - siehe das Anhang-Kapitel Lineare gemischte Modelle.
Daten
Dies ist derselbe Versuch wie im vorherigen Kapitel, erneut aus Gomez und Gomez (1984): ein Ertragsversuch (kg/ha), der 4 Genotypen (G) mit 6 Stickstoffstufen (N) kreuzt. Der einzige Unterschied ist die Versuchsanlage - und dementsprechend eine zusätzliche Spalte mainplot, die die 18 Großparzellen identifiziert.
Hinweis
Die yield-Werte in diesem Datensatz sind identisch mit denen im vorherigen Kapitel; nur die räumliche Anordnung (und damit die mainplot-Spalte) unterscheidet sich. Das erlaubt uns, an genau denselben Zahlen zu sehen, wie das angenommene Design das Modell und die Inferenz verändert.
Import
Rows: 72 Columns: 7
── Column specification ────────────────────────────────────────────────────────
Delimiter: ","
chr (4): rep, mainplot, G, N
dbl (3): yield, row, col
ℹ Use `spec()` to retrieve the full column specification for this data.
ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.# A tibble: 72 × 7
yield row col rep mainplot G N
<dbl> <dbl> <dbl> <chr> <chr> <chr> <chr>
1 4520 4 1 rep1 mp01 Simba Goomba
2 5598 2 2 rep1 mp02 Simba Koopa
3 6192 1 3 rep1 mp03 Simba Toad
4 8542 2 4 rep1 mp04 Simba Peach
5 5806 2 5 rep1 mp05 Simba Diddy
6 7470 1 6 rep1 mp06 Simba Yoshi
7 4034 2 1 rep1 mp01 Nala Goomba
8 6682 4 2 rep1 mp02 Nala Koopa
9 6869 3 3 rep1 mp03 Nala Toad
10 6318 4 4 rep1 mp04 Nala Peach
# ℹ 62 more rowsDer Datensatz enthält:
-
N: Sechs Stickstoffstufen - der Großparzellen-Faktor -
G: Vier Genotypen - der Unterparzellen-Faktor -
rep: Drei vollständige Blöcke -
mainplot: Die 18 Großparzellen (6 je Block) -
yield: Ernteertrag in kg/ha -
rowundcol: Feldparzellenkoordinaten für die Visualisierung mit desplot
Format
Wir codieren den Block, die Großparzellen-Kennung und beide Behandlungsfaktoren als Faktoren:
# A tibble: 72 × 7
yield row col rep mainplot G N
<dbl> <dbl> <dbl> <fct> <fct> <fct> <fct>
1 4520 4 1 rep1 mp01 Simba Goomba
2 5598 2 2 rep1 mp02 Simba Koopa
3 6192 1 3 rep1 mp03 Simba Toad
4 8542 2 4 rep1 mp04 Simba Peach
5 5806 2 5 rep1 mp05 Simba Diddy
6 7470 1 6 rep1 mp06 Simba Yoshi
7 4034 2 1 rep1 mp01 Nala Goomba
8 6682 4 2 rep1 mp02 Nala Koopa
9 6869 3 3 rep1 mp03 Nala Toad
10 6318 4 4 rep1 mp04 Nala Peach
# ℹ 62 more rowsErkunden
Wie zuvor beginnen wir mit einfaktoriellen Zusammenfassungen des Ertrags:
# A tibble: 6 × 4
N count mean_yield sd_yield
<fct> <int> <dbl> <dbl>
1 Diddy 12 5866. 832.
2 Toad 12 5864. 1434.
3 Yoshi 12 5812 2349.
4 Peach 12 5797. 2660.
5 Koopa 12 5478. 657.
6 Goomba 12 4054. 672.# A tibble: 4 × 4
G count mean_yield sd_yield
<fct> <int> <dbl> <dbl>
1 Simba 18 6554. 1475.
2 Nala 18 6156. 1078.
3 Timon 18 5563. 1269.
4 Pumba 18 3642. 1434.Da dies dieselben Ertragswerte wie zuvor sind, stimmen die Zusammenfassungen mit denen im vorherigen Kapitel überein. Wir definieren wieder eine feste Palette für die sechs Stickstoffstufen und tragen den Ertrag gegen Stickstoff auf, ein Panel je Genotyp:
ggplot(data = dat) +
aes(y = yield, x = N, color = N) +
facet_wrap(~G, labeller = label_both) +
stat_summary(
fun = mean,
colour = "grey",
geom = "line",
linetype = "dotted",
group = 1
) +
geom_point() +
scale_x_discrete(name = "Stickstoff") +
scale_y_continuous(
name = "Ertrag",
limits = c(0, NA),
expand = expansion(mult = c(0, 0.1))
) +
scale_color_manual(values = Ncolors, guide = "none") +
theme_bw() +
theme(axis.text.x = element_text(angle = 45, hjust = 1, vjust = 1))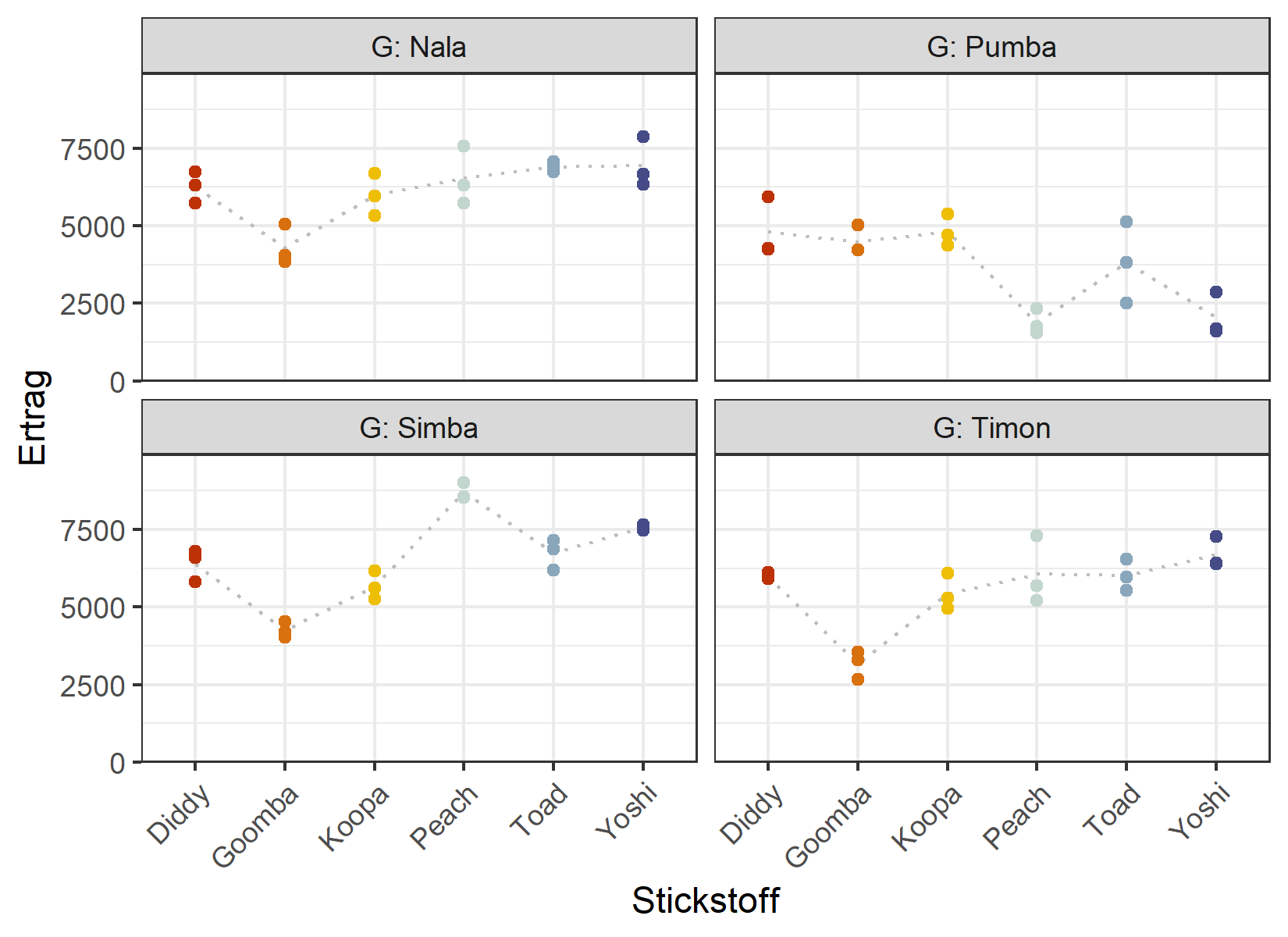
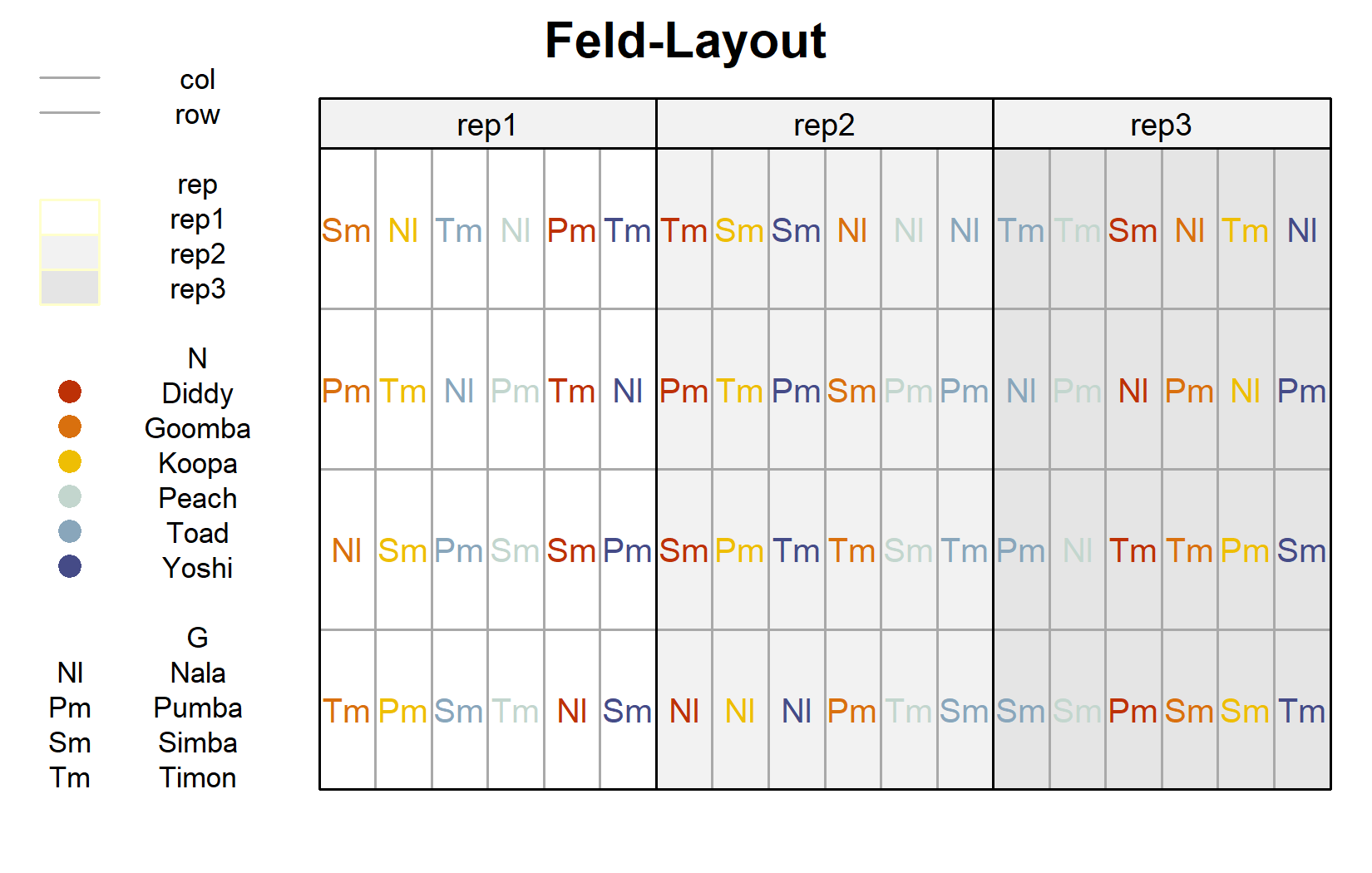
Beim Feld-Layout wird die Split-Plot-Struktur sichtbar. Wir zeichnen zunächst das übliche Layout (Genotyp-Beschriftungen nach Stickstoffstufe gefärbt) und heben dann die Großparzellen hervor:
desplot(
data = dat,
form = rep ~ col + row | rep, # ein Panel je Block
col.regions = c("white", "grey95", "grey90"),
text = G, # Genotypnamen je Parzelle
cex = 0.8, # Genotypnamen: Schriftgröße
shorten = "abb", # Genotypnamen: abkürzen
col = N, # Genotypnamen nach Stickstoffstufe färben
col.text = Ncolors, # die eigenen Stickstofffarben verwenden
out1 = col, out1.gpar = list(col = "darkgrey"), # Linien zwischen Spalten
out2 = row, out2.gpar = list(col = "darkgrey"), # Linien zwischen Reihen
main = "Feld-Layout",
show.key = TRUE,
key.cex = 0.7
)
# eine eigene Farbe je Großparzelle (18 insgesamt)
mainplotcolors <- c(met.brewer("VanGogh3", 6),
met.brewer("Hokusai2", 6),
met.brewer("OKeeffe2", 6)) %>%
as.vector() %>%
set_names(levels(dat$mainplot))
desplot(
data = dat,
form = mainplot ~ col + row | rep, # Farbe je Großparzelle, ein Panel je Block
col.regions = mainplotcolors,
out1 = col, out1.gpar = list(col = "darkgrey"),
out2 = row, out2.gpar = list(col = "darkgrey"),
main = "Großparzellen",
show.key = FALSE
)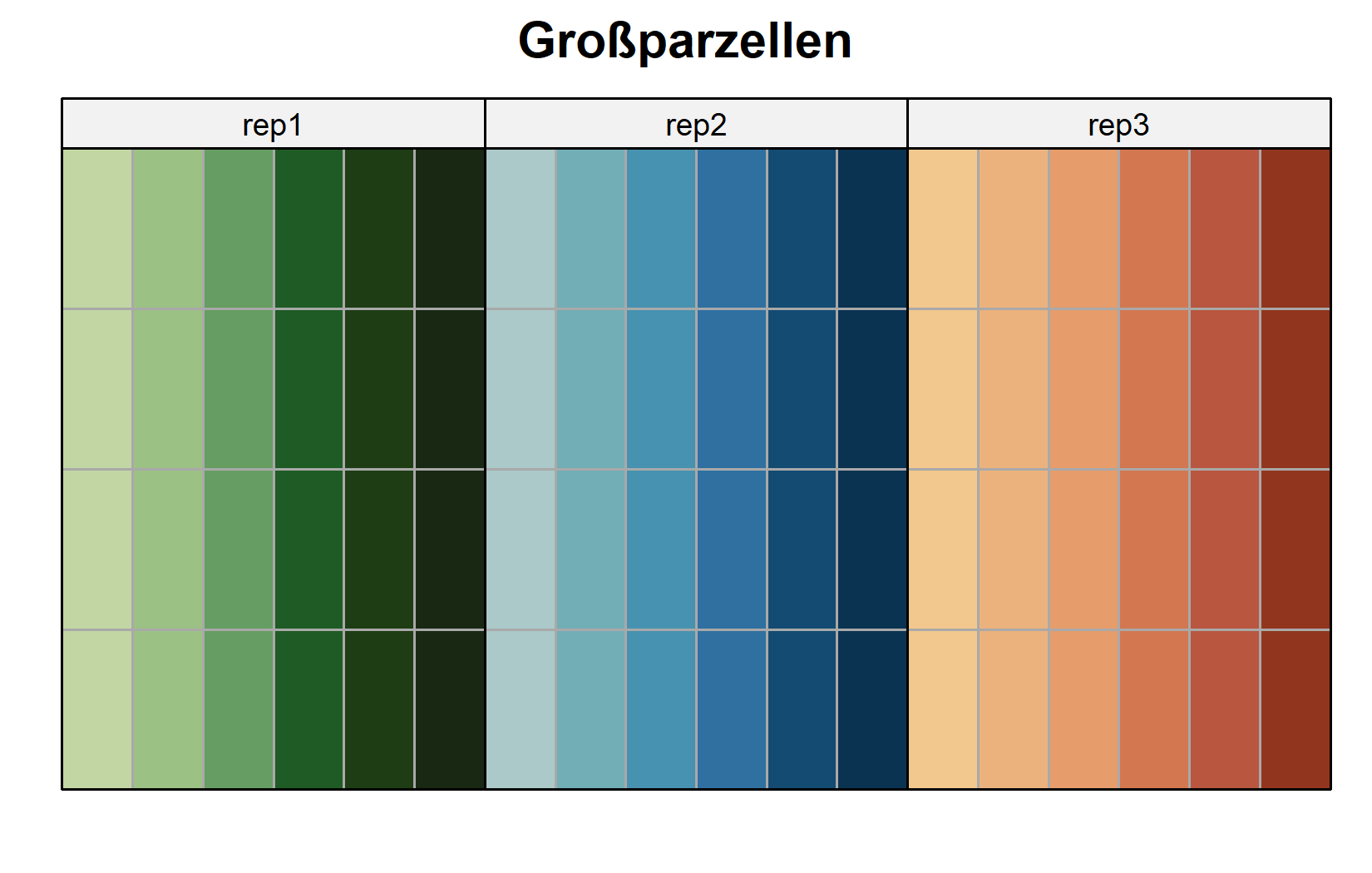
Das zweite Layout macht das Design explizit: Jeder gefärbte Block von Zellen ist eine Großparzelle, trägt eine einzige Stickstoffstufe und ist intern in die vier Genotyp-Unterparzellen aufgeteilt. Die Stickstoffstufen werden zwischen den Großparzellen randomisiert, die Genotypen innerhalb von ihnen.
Modell
Der Behandlungsteil des Modells ist derselbe wie im RCBD-Kapitel: die beiden Behandlungsfaktoren G und N als Haupteffekte plus ihre Interaktion G:N und der Blockeffekt rep. Neu ist der Zufallseffekt für die Großparzellen, (1 | rep:mainplot), der die 18 Großparzellen als zusätzliche Randomisierungsebene repräsentiert:
mod <- lmer(yield ~ G + N + G:N + rep + (1 | rep:mainplot),
data = dat)Die Syntax (1 | rep:mainplot) weist lmer() an, die Kombination von rep und mainplot (d.h. die 18 eindeutigen Großparzellen) als Zufallseffekt zu behandeln. Das ist der einzige strukturelle Unterschied zum RCBD-Modell lm(yield ~ N + G + N:G + rep) des vorherigen Kapitels.
WarnungModellannahmen erfüllt?
An dieser Stelle (d.h. nach dem Modell-Fit und vor der ANOVA-Interpretation) sollte man prüfen, ob die Modellannahmen erfüllt sind. Mehr dazu im Anhang A1: Modelldiagnostik.
ANOVA
Für gemischte Modelle verwenden wir eine ANOVA mit Kenward-Roger-Freiheitsgraden, die genauere F-Tests in den für geplante Versuche typischen Situationen mit kleinen Stichproben liefert:
ANOVA <- anova(mod, ddf = "Kenward-Roger")
ANOVAType III Analysis of Variance Table with Kenward-Roger's method
Sum Sq Mean Sq NumDF DenDF F value Pr(>F)
G 89885051 29961684 3 36 85.7416 < 2.2e-16 ***
N 19192886 3838577 5 10 10.9849 0.0008277 ***
rep 683088 341544 2 10 0.9774 0.4095330
G:N 69378044 4625203 15 36 13.2360 2.078e-10 ***
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1Die Interaktion G:N ist statistisch signifikant, genau wie in der RCBD-Analyse. Der lehrreichste Teil dieser Tabelle sind jedoch die Nenner-Freiheitsgrade (DenDF):
- Der Großparzellen-Faktor
Nwird gegen nur 10 Nenner-Freiheitsgrade getestet, weil er auf der Ebene der Großparzellen verglichen wird, von denen es wenige gibt. - Der Unterparzellen-Faktor
Gund die InteraktionG:Nwerden gegen 36 Nenner-Freiheitsgrade getestet, weil sie auf der feineren Unterparzellen-Ebene verglichen werden.
Das ist das Kennzeichen eines Split-Plot-Designs: Der Großparzellen-Faktor wird weniger präzise geschätzt als der Unterparzellen-Faktor und die Interaktion. Wir kommen darauf zurück, wenn wir die beiden Analysen unten vergleichen.
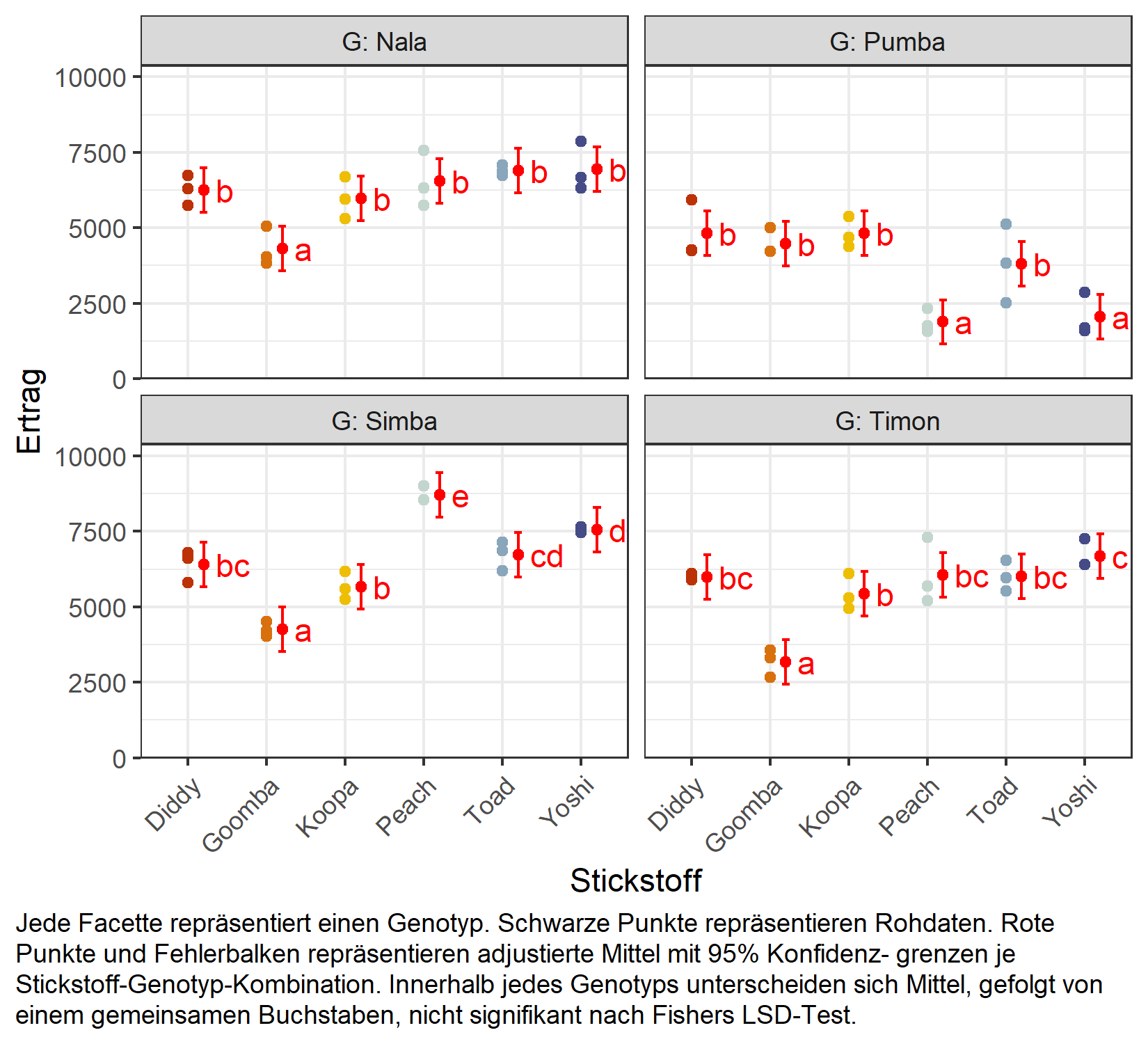
Mittelwertvergleich
Wegen der signifikanten Interaktion vergleichen wir erneut die Stickstoffstufen innerhalb jedes Genotyps über specs = ~ N | G:
G = Nala:
N emmean SE df lower.CL upper.CL .group
Goomba 4306 366 41.9 3568 5044 a
Koopa 5982 366 41.9 5244 6720 b
Diddy 6259 366 41.9 5521 6997 b
Peach 6540 366 41.9 5803 7278 b
Toad 6895 366 41.9 6157 7633 b
Yoshi 6951 366 41.9 6213 7688 b
G = Pumba:
N emmean SE df lower.CL upper.CL .group
Peach 1881 366 41.9 1143 2618 a
Yoshi 2047 366 41.9 1309 2784 a
Toad 3816 366 41.9 3078 4554 b
Goomba 4481 366 41.9 3744 5219 b
Diddy 4812 366 41.9 4074 5550 b
Koopa 4816 366 41.9 4078 5554 b
G = Simba:
N emmean SE df lower.CL upper.CL .group
Goomba 4253 366 41.9 3515 4990 a
Koopa 5672 366 41.9 4934 6410 b
Diddy 6400 366 41.9 5662 7138 bc
Toad 6733 366 41.9 5995 7470 cd
Yoshi 7563 366 41.9 6826 8301 d
Peach 8701 366 41.9 7963 9438 e
G = Timon:
N emmean SE df lower.CL upper.CL .group
Goomba 3177 366 41.9 2440 3915 a
Koopa 5443 366 41.9 4705 6180 b
Diddy 5994 366 41.9 5256 6732 bc
Toad 6014 366 41.9 5276 6752 bc
Peach 6065 366 41.9 5328 6803 bc
Yoshi 6687 366 41.9 5950 7425 c
Results are averaged over the levels of: rep
Degrees-of-freedom method: kenward-roger
Confidence level used: 0.95
significance level used: alpha = 0.05
NOTE: If two or more means share the same grouping symbol,
then we cannot show them to be different.
But we also did not show them to be the same. Wie im vorherigen Kapitel bleiben die Vergleiche unadjustiert (adjust = "none", d.h. Fishers LSD); siehe Anhang A4: Multiplizitätskorrekturen für die Alternativen und Anhang A5: Compact Letter Display für die Buchstabendarstellung. Man beachte, dass die Standardfehler nun aus dem gemischten Modell stammen und daher die Split-Plot-Fehlerstruktur korrekt widerspiegeln.
my_caption <- "Jede Facette repräsentiert einen Genotyp. Schwarze Punkte repräsentieren
Rohdaten. Rote Punkte und Fehlerbalken repräsentieren adjustierte Mittel mit 95% Konfidenz-
grenzen je Stickstoff-Genotyp-Kombination. Innerhalb jedes Genotyps unterscheiden sich Mittel,
gefolgt von einem gemeinsamen Buchstaben, nicht signifikant nach Fishers LSD-Test."
ggplot() +
facet_wrap(~G, labeller = label_both) +
aes(x = N) +
# schwarze Punkte für die Rohdaten
geom_point(
data = dat,
aes(y = yield, color = N)
) +
# rote Punkte für die adjustierten Mittel
geom_point(
data = mean_comp,
aes(y = emmean),
color = "red",
position = position_nudge(x = 0.2)
) +
# rote Fehlerbalken für die Konfidenzgrenzen der adjustierten Mittel
geom_errorbar(
data = mean_comp,
aes(ymin = lower.CL, ymax = upper.CL),
color = "red",
width = 0.1,
position = position_nudge(x = 0.2)
) +
# rote Buchstaben
geom_text(
data = mean_comp,
aes(y = emmean, label = str_trim(.group)),
color = "red",
position = position_nudge(x = 0.35),
hjust = 0
) +
scale_x_discrete(name = "Stickstoff") +
scale_y_continuous(
name = "Ertrag",
limits = c(0, NA),
expand = expansion(mult = c(0, 0.1))
) +
scale_color_manual(values = Ncolors, guide = "none") +
theme_bw() +
labs(caption = my_caption) +
theme(
plot.caption = element_textbox_simple(margin = margin(t = 5)),
plot.caption.position = "plot",
axis.text.x = element_text(angle = 45, hjust = 1, vjust = 1)
)
RCBD vs. Split-Plot: Was hat sich geändert?
Da die Ertragswerte mit denen im vorherigen Kapitel identisch sind, können wir die beiden Analysen direkt vergleichen und den Effekt der Design-Annahme allein isolieren.
-
Die Behandlungsterme sind dieselben. Beide Modelle enthalten
G,N,G:Nundrep. Die Punktschätzer der Mittel sind im Wesentlichen unverändert. -
Die Fehlerstruktur unterscheidet sich. Das RCBD-Modell hat einen einzigen Residualfehler (
lm()); das Split-Plot-Modell fügt einen zufälligen Großparzellen-Effekt(1 | rep:mainplot)hinzu, der die Variation in ein Großparzellen-Stratum und ein Unterparzellen-Stratum aufteilt. -
Die Konsequenz ist Präzision. In der RCBD-Analyse wurde der Stickstoff-Haupteffekt gegen den großen Residualfehler getestet (viele Freiheitsgrade). In der Split-Plot-Analyse wird er gegen den Großparzellen-Fehler getestet (nur 10 Freiheitsgrade). Die Evidenz für einen Stickstoffeffekt ist daher hier merklich schwächer - nicht weil sich die Daten geändert haben, sondern weil ein Split-Plot anerkennt, dass Stickstoff nicht unabhängig jeder Parzelle zugewiesen wurde. Umgekehrt werden der Unterparzellen-Faktor
Gund die InteraktionG:Nmit guter Präzision geschätzt.
Die Kernaussage ist, dass das Design die Analyse bestimmt. Einen Split-Plot-Versuch so zu behandeln, als wäre er ein vollständig randomisiertes RCBD, würde die Präzision des Großparzellen-Faktors überschätzen und könnte zu falsch-positiven Schlüssen über ihn führen.
Abschluss
Wir haben nun unser erstes praktisches gemischtes Modell gefittet und gesehen, wie ein Split-Plot-Design Großparzellen- von Unterparzellen-Information trennt. Die Behandlungsstruktur sah genau wie beim zweifaktoriellen RCBD aus, aber ein einziger Zufallseffekt-Term veränderte die Inferenz auf eine Weise, die genau widerspiegelt, wie der Versuch tatsächlich durchgeführt wurde.
HinweisZusammenfassung
Ein Split-Plot-Design hat zwei Ebenen von Versuchseinheiten: Großparzellen (hier: Stickstoff) und Unterparzellen (hier: Genotypen), mit einer eigenen Randomisierung auf jeder Ebene.
Zwei Randomisierungen erfordern ein gemischtes Modell. Die Großparzellen gehen als Zufallseffekt ein:
yield ~ G + N + G:N + rep + (1 | rep:mainplot), gefittet mitlmer().Kenward-Roger-Freiheitsgrade werden für die ANOVA des gemischten Modells verwendet.
Großparzellen-Faktoren werden weniger präzise getestet als Unterparzellen-Faktoren und die Interaktion - sichtbar an den deutlich kleineren Nenner-Freiheitsgraden für
N.Dieselben Daten, anderes Design, andere Inferenz. Verglichen mit der RCBD-Analyse der identischen Erträge spiegelt das Split-Plot die Struktur des Versuchs korrekt wider und verändert die Präzision des Großparzellen-Faktors.
Literatur
Gomez, Kwanchai A, und Arturo A Gomez. 1984. Statistical procedures for agricultural research. 2. Aufl. An International Rice Research Institute book. Nashville, TN: John Wiley & Sons.
Zitat
Mit BibTeX zitieren:
@online{schmidt2026,
author = {{Dr. Paul Schmidt}},
publisher = {BioMath GmbH},
title = {2. Zweifaktorielle ANOVA in einem Split-Plot-Design},
date = {2026-06-08},
url = {https://biomathcontent.netlify.app/de/content/lin_mod_adv/02_splitplot.html},
langid = {de}
}
Bitte zitieren Sie diese Arbeit als:
Dr. Paul Schmidt. 2026. “2. Zweifaktorielle ANOVA in einem
Split-Plot-Design.” BioMath GmbH. June 8, 2026. https://biomathcontent.netlify.app/de/content/lin_mod_adv/02_splitplot.html.