for (pkg in c("agridat", "desplot", "emmeans", "ggtext", "here", "lme4",
"lmerTest", "multcomp", "multcompView", "tidyverse")) {
if (!require(pkg, character.only = TRUE)) install.packages(pkg)
}
library(agridat)
library(desplot)
library(emmeans)
library(ggtext)
library(here)
library(lme4)
library(lmerTest)
library(multcomp)
library(multcompView)
library(tidyverse)To install and load all the packages used in this chapter, run the following code:
Row-Column Designs
In Chapter 3, we encountered the Latin Square design, which controls for two sources of variation (rows and columns) simultaneously. However, the Latin Square has a major limitation: the number of treatments must equal the number of rows and columns. This makes it impractical for experiments with many treatments.
What is a Row-Column Design?
A resolvable row-column design extends the Latin Square concept to accommodate more treatments. Like an alpha design, it has complete replicates that are subdivided - but here each replicate is subdivided into both incomplete rows and incomplete columns. This provides double blocking within each replicate.
The key features are:
- Two-dimensional blocking: Each replicate has both row and column structure
- Incomplete blocks: Neither rows nor columns contain all treatments
- Resolvability: Replicates are complete, containing each treatment exactly once
- Flexibility: Can accommodate various numbers of treatments
The advantages include:
- Control of two gradients: Accounts for spatial trends in two directions simultaneously
- More treatments than Latin Square: Not limited to t×t arrangements
- Increased precision: Double blocking can substantially reduce experimental error
- Practical for field trials: Matches the rectangular layout of many field experiments
Data
This example considers data published in Kempton, Fox, and Cerezo (1997) from a yield trial laid out as a resolvable row-column design. The trial had 35 genotypes (gen), 2 complete replicates (rep) with 5 rows (row) and 7 columns (col) each. Thus, each replicate forms a 5×7 grid with incomplete rows and columns.
Import
The data is available as part of the {agridat} package:
dat <- as_tibble(agridat::kempton.rowcol)
dat# A tibble: 68 × 5
rep row col gen yield
<fct> <int> <int> <fct> <dbl>
1 R1 1 1 G20 3.77
2 R1 1 2 G04 3.21
3 R1 1 3 G33 4.55
4 R1 1 4 G28 4.09
5 R1 1 5 G07 5.05
6 R1 1 6 G12 4.19
7 R1 1 7 G30 3.27
8 R1 2 1 G10 3.44
9 R1 2 2 G14 4.3
10 R1 2 4 G21 3.86
# ℹ 58 more rowsThe dataset contains:
-
rep: Two complete replicates (R1, R2) -
row: Row position within replicate (1-5) -
col: Column position within replicate (1-7) -
gen: 35 genotypes -
yield: Crop yield
Note that there are missing values in this dataset - two plots have no yield recorded.
Format
For our analysis, gen should be encoded as a factor. We also create factor versions of row and col for the statistical model:
# A tibble: 68 × 7
rep row col gen yield rowF colF
<fct> <int> <int> <fct> <dbl> <fct> <fct>
1 R1 1 1 G20 3.77 1 1
2 R1 1 2 G04 3.21 1 2
3 R1 1 3 G33 4.55 1 3
4 R1 1 4 G28 4.09 1 4
5 R1 1 5 G07 5.05 1 5
6 R1 1 6 G12 4.19 1 6
7 R1 1 7 G30 3.27 1 7
8 R1 2 1 G10 3.44 2 1
9 R1 2 2 G14 4.3 2 2
10 R1 2 4 G21 3.86 2 4
# ℹ 58 more rowsExplore
Let’s examine the summary statistics by genotype:
# A tibble: 35 × 4
gen count mean_yield sd_yield
<fct> <int> <dbl> <dbl>
1 G19 2 6.07 1.84
2 G07 2 5.74 0.976
3 G33 2 5.13 0.820
4 G06 2 4.96 0.940
5 G09 2 4.94 1.68
6 G11 2 4.93 1.03
7 G14 2 4.92 0.877
8 G27 2 4.89 1.80
9 G03 2 4.78 0.0424
10 G25 2 4.78 0.361
# ℹ 25 more rowsMost genotypes appear twice (once per replicate), but some have only one observation due to missing data. Let’s visualize the data:
# sort genotypes by mean yield
gen_order <- dat %>%
group_by(gen) %>%
summarise(mean = mean(yield, na.rm = TRUE)) %>%
arrange(mean) %>%
pull(gen) %>%
as.character()
ggplot(data = dat) +
aes(
y = yield,
x = gen,
shape = rep
) +
geom_line(
aes(group = gen),
color = "darkgrey"
) +
geom_point() +
scale_x_discrete(
name = "Genotype",
limits = gen_order
) +
scale_y_continuous(
name = "Yield",
limits = c(0, NA),
expand = expansion(mult = c(0, 0.05))
) +
scale_shape_discrete(
name = "Replicate"
) +
guides(shape = guide_legend(nrow = 1)) +
theme_classic() +
theme(
legend.position = "top",
axis.text.x = element_text(angle = 90, vjust = 0.5)
)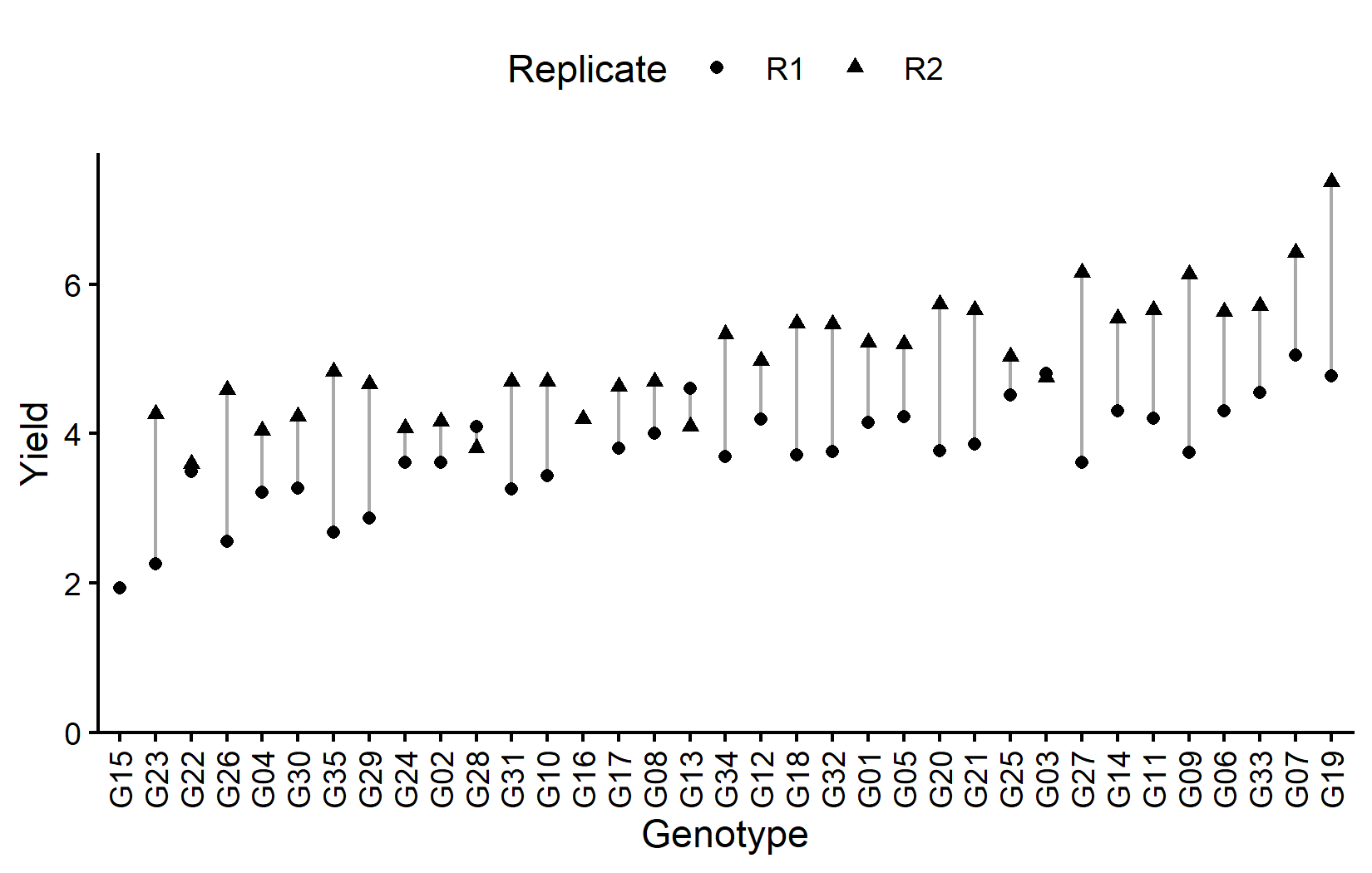
Now let’s look at the field layout. Note that the two missing plots will appear as white/empty:
desplot(
data = dat,
form = gen ~ col + row | rep, # fill color per genotype, panels per replicate
text = gen,
cex = 0.7,
shorten = FALSE,
out1 = row, out1.gpar = list(col = "black"), # lines between rows
out2 = col, out2.gpar = list(col = "black"), # lines between columns
main = "Field layout",
show.key = FALSE
) 
The black lines show the row and column structure within each replicate. Each genotype appears once per replicate, but in different row-column positions.
Model and ANOVA
Choosing Between Fixed and Random Effects
For a row-column design, the model needs to account for row and column effects within each replicate. We can treat these as either fixed or random effects. Let’s compare both approaches:
Now compare the average s.e.d. for genotype comparisons:
NOTE: A nesting structure was detected in the fitted model:
rowF %in% rep, colF %in% rep# A tibble: 2 × 2
model mean_sed
<chr> <dbl>
1 Fixed row/col 0.408
2 Random row/col 0.402In this case, the fixed effects model has a slightly smaller s.e.d., so we’ll use it for our analysis.
WarningModel assumptions met?
It is at this point (i.e. after fitting the model and before interpreting the ANOVA) that one should check whether the model assumptions are met. Find out more in Appendix A1: Model Diagnostics.
Conducting the ANOVA
ANOVA <- anova(mod_fixed)
ANOVAAnalysis of Variance Table
Response: yield
Df Sum Sq Mean Sq F value Pr(>F)
gen 34 32.157 0.9458 10.7456 4.969e-05 ***
rep 1 24.901 24.9014 282.9193 1.042e-09 ***
rep:rowF 8 2.512 0.3140 3.5680 0.023647 *
rep:colF 12 6.327 0.5273 5.9905 0.002067 **
Residuals 12 1.056 0.0880
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1The genotype effect is not statistically significant (p > 0.05), indicating no strong evidence for differences among genotypes in this trial. However, let’s still examine the mean comparisons.
Mean Comparisons
gen emmean SE df lower.CL upper.CL .group
G04 3.48 0.270 12 2.89 4.07 a
G23 3.58 0.270 12 2.99 4.17 ab
G15 3.60 0.442 12 2.64 4.57 abcde
G35 3.63 0.277 12 3.03 4.24 abc
G31 3.79 0.280 12 3.18 4.40 abcd
G02 3.84 0.279 12 3.23 4.45 abcd
G26 3.90 0.291 12 3.26 4.53 abcde
G24 3.90 0.267 12 3.32 4.49 abcd f
G29 3.91 0.276 12 3.31 4.51 abcde
G30 3.99 0.270 12 3.40 4.58 abcdefg
G32 4.12 0.276 12 3.52 4.72 abcdefgh
G17 4.14 0.282 12 3.53 4.75 abcdefgh
G09 4.15 0.274 12 3.56 4.75 abcdefgh
G34 4.20 0.267 12 3.62 4.78 abcdefgh
G16 4.23 0.432 12 3.29 5.17 abcdefghij
G05 4.25 0.278 12 3.64 4.85 abcdefghi
G20 4.25 0.266 12 3.67 4.83 abcdefgh
G22 4.27 0.282 12 3.66 4.88 abcdefghi
G10 4.36 0.278 12 3.76 4.97 abcdefghij
G28 4.37 0.278 12 3.77 4.98 bcdefghij
G18 4.48 0.284 12 3.86 5.10 cdefghij
G21 4.57 0.269 12 3.98 5.16 defghij
G08 4.58 0.285 12 3.95 5.20 defghij
G25 4.59 0.277 12 3.98 5.19 defghij
G13 4.73 0.284 12 4.11 5.35 e ghijkl
G27 4.75 0.282 12 4.13 5.36 fghijk
G33 4.76 0.286 12 4.13 5.38 e ghijk
G14 4.79 0.270 12 4.20 5.38 ghijkl
G01 4.88 0.268 12 4.30 5.46 hijkl
G07 4.94 0.270 12 4.35 5.53 hijkl
G11 4.97 0.276 12 4.37 5.57 hijkl
G12 5.13 0.293 12 4.49 5.77 ijkl
G03 5.15 0.281 12 4.54 5.76 jkl
G06 5.53 0.280 12 4.92 6.14 kl
G19 5.60 0.281 12 4.99 6.22 l
Results are averaged over the levels of: colF, rowF, rep
Confidence level used: 0.95
significance level used: alpha = 0.05
NOTE: If two or more means share the same grouping symbol,
then we cannot show them to be different.
But we also did not show them to be the same. The compact letter display shows the groupings based on Fisher’s LSD test. With 35 genotypes, the letter display becomes complex, but it still provides a concise summary of which genotypes differ significantly.
Visualizing Results
my_caption <- "Black dots represent raw data. Red diamonds and error bars represent adjusted means with 95% confidence limits per genotype. Means followed by a common letter are not significantly different according to Fisher's LSD test."
ggplot() +
aes(x = gen) +
# black dots representing the raw data
geom_point(
data = dat,
aes(y = yield)
) +
# red diamonds representing the adjusted means
geom_point(
data = mean_comp,
aes(y = emmean),
shape = 18,
color = "red",
position = position_nudge(x = 0.2)
) +
# red error bars representing the confidence limits of the adjusted means
geom_errorbar(
data = mean_comp,
aes(ymin = lower.CL, ymax = upper.CL),
color = "red",
width = 0.1,
position = position_nudge(x = 0.2)
) +
# red letters
geom_text(
data = mean_comp,
aes(y = upper.CL, label = str_trim(.group)),
color = "red",
angle = 90,
hjust = -0.2,
position = position_nudge(x = 0.2)
) +
scale_x_discrete(
name = "Genotype",
limits = as.character(mean_comp$gen)
) +
scale_y_continuous(
name = "Yield",
expand = expansion(mult = c(0, 0.05))
) +
coord_cartesian(ylim = c(0, NA)) +
labs(caption = my_caption) +
theme_classic() +
theme(plot.caption = element_textbox_simple(margin = margin(t = 5)),
plot.caption.position = "plot",
axis.text.x = element_text(angle = 90, vjust = 0.5))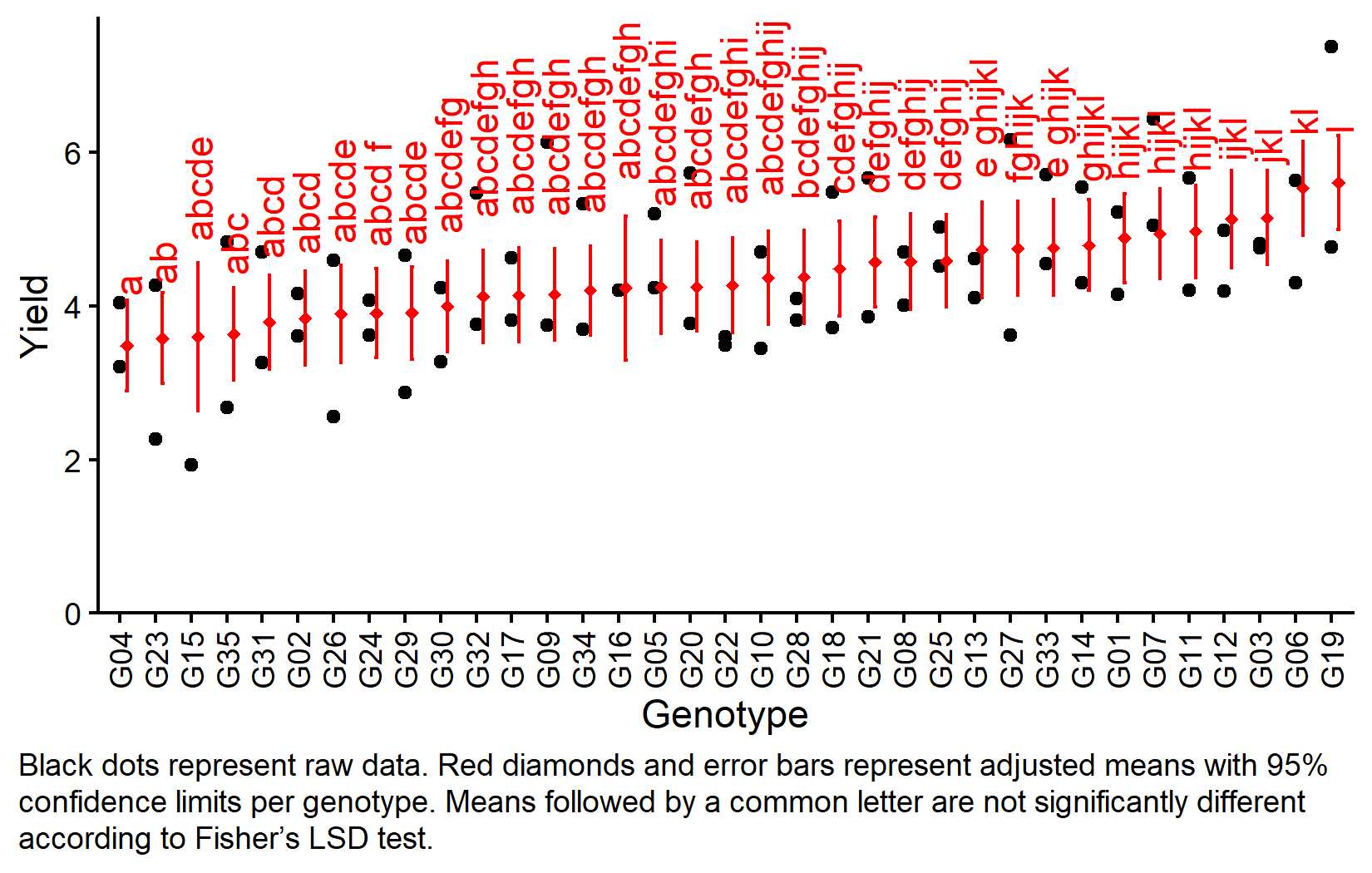
Bonus: Design Efficiency
Let’s assess the efficiency of the row-column design compared to a simple RCBD (ignoring the row and column structure):
[1] 1.953932An efficiency > 1 indicates that the row-column design is more efficient than a simple RCBD, meaning the row and column blocking successfully reduced experimental error.
Wrapping Up
You’ve now learned how to analyze data from a resolvable row-column design, which provides powerful control over two sources of spatial variation.
NoteKey Takeaways
Row-column designs control for two sources of variation simultaneously by blocking in both row and column directions within each replicate.
More flexible than Latin Square: Can accommodate any number of treatments, not limited to t×t arrangements.
Double blocking within replicates provides increased precision when spatial trends exist in two directions.
Model choice between fixed and random row/column effects can be based on which gives smaller average s.e.d.
The model with fixed effects:
yield ~ gen + rep + rep:rowF + rep:colF(rows and columns nested within replicates).Design efficiency > 1 compared to RCBD confirms the benefit of the additional row-column structure.
Missing data handling: Row-column designs can still be analyzed when some observations are missing, though precision may be affected.
Design Comparison Summary
Let’s summarize the progression of designs covered in this chapter series:
| Design | Blocking Structure | Model Formula | Best For |
|---|---|---|---|
| CRD | None | y ~ trt |
Homogeneous conditions |
| RCBD | Complete blocks | y ~ trt + block |
One gradient |
| Latin Square | Rows + columns (complete) | y ~ trt + row + col |
Two gradients, few treatments |
| Alpha Design | Incomplete blocks in reps | y ~ trt + rep + (1|rep:block) |
Many treatments, one gradient |
| Augmented | Checks + entries | y ~ trt + block |
Screening many unreplicated entries |
| Row-Column | Inc. rows + cols in reps | y ~ trt + rep + rep:row + rep:col |
Many treatments, two gradients |
References
Kempton, R. A., P. N. Fox, and M. Cerezo. 1997. Statistical Methods for Plant Variety Evaluation. Springer Netherlands. https://doi.org/10.1007/978-94-009-1503-9.
Citation
BibTeX citation:
@online{schmidt2026,
author = {{Dr. Paul Schmidt}},
publisher = {BioMath GmbH},
title = {6. {One-way} {ANOVA} in a {Row-Column} {Design}},
date = {2026-03-04},
url = {https://biomathcontent.netlify.app/content/lin_mod_exp/06_oneway_rowcol.html},
langid = {en}
}
For attribution, please cite this work as:
Dr. Paul Schmidt. 2026. “6. One-Way ANOVA in a Row-Column
Design.” BioMath GmbH. March 4, 2026. https://biomathcontent.netlify.app/content/lin_mod_exp/06_oneway_rowcol.html.