for (pkg in c("desplot", "emmeans", "ggtext", "here", "lme4",
"lmerTest", "multcomp", "multcompView", "tidyverse")) {
if (!require(pkg, character.only = TRUE)) install.packages(pkg)
}
library(desplot)
library(emmeans)
library(ggtext)
library(here)
library(lme4)
library(lmerTest)
library(multcomp)
library(multcompView)
library(tidyverse)Um alle in diesem Kapitel verwendeten Pakete zu installieren und zu laden, führen Sie folgenden Code aus:
Augmented Designs
Im vorherigen Kapitel haben wir ein Alpha-Design analysiert, bei dem alle Genotypen über Blöcke hinweg repliziert wurden. In der Pflanzenzüchtung und Sortenprüfung stehen wir jedoch oft vor Situationen, in denen wir viele neue Genotypen testen müssen, aber nur begrenzte Ressourcen haben. Die Prüfung aller Genotypen mit vollständiger Replikation ist möglicherweise nicht durchführbar.
Was ist ein Augmented Design?
Ein Augmented Design (auch erweitertes Blockdesign genannt) adressiert dies durch die Aufnahme von zwei Arten von Einträgen:
- Standardsorten (Checks): Repliziert über alle Blöcke, bieten eine Basis für die Schätzung von Blockeffekten
- Neue Einträge (Prüfgenotypen): Nicht repliziert, erscheinen nur in jeweils einem Block
Die replizierten Standards ermöglichen es uns, Blockeffekte zu schätzen und zu adjustieren, die dann auf die nicht replizierten Einträge angewendet werden können. Dieses Design maximiert die Anzahl neuer Einträge, die mit begrenzten Ressourcen getestet werden können, und ermöglicht dennoch valide statistische Vergleiche.
Die Vorteile von Augmented Designs umfassen:
- Ressourceneffizienz: Viele neue Einträge ohne vollständige Replikation testen
- Valide Vergleiche: Aus Standards geschätzte Blockeffekte werden auf alle Einträge angewendet
- Flexibilität: Kann unterschiedliche Anzahlen neuer Einträge pro Block aufnehmen
- Praktisch für Screening: Ideal für frühe Sortenprüfungen mit vielen Kandidaten
Der Kompromiss
Der wesentliche Kompromiss ist die Präzision: Nicht replizierte Einträge haben höhere Standardfehler als replizierte Standards. Das bedeutet, dass Vergleiche mit neuen Einträgen weniger präzise sind als Vergleiche zwischen Standards. Für anfängliche Screening-Zwecke ist dies jedoch oft akzeptabel.
Daten
Dieses Beispiel betrachtet Daten, die in Petersen (1994) veröffentlicht wurden, aus einem Ertragsversuch, der als Augmented Design angelegt wurde. Der Versuch umfasste 3 Standardsorten (st, ci, wa), die in allen 6 Blöcken repliziert wurden, und 30 neue Einträge (nummeriert 1-30), die jeweils nur in einem Block erschienen.
Import
Rows: 48 Columns: 5
── Column specification ────────────────────────────────────────────────────────
Delimiter: ","
chr (2): gen, block
dbl (3): yield, row, col
ℹ Use `spec()` to retrieve the full column specification for this data.
ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.# A tibble: 48 × 5
gen yield block row col
<chr> <dbl> <chr> <dbl> <dbl>
1 st 2972 I 1 1
2 14 2405 I 2 1
3 26 2855 I 3 1
4 ci 2592 I 4 1
5 17 2572 I 5 1
6 wa 2608 I 6 1
7 22 2705 I 7 1
8 13 2391 I 8 1
9 st 3122 II 1 2
10 ci 3023 II 2 2
# ℹ 38 more rowsDer Datensatz enthält:
-
gen: Genotypbezeichnung (3 Standards: st, ci, wa; 30 neue Einträge: 1-30) -
yield: Ernteertrag -
block: Sechs Blöcke (I-VI) -
rowundcol: Feldparzellenkoordinaten für die Visualisierung
Formatierung
Vor der Analyse müssen wir gen und block als Faktoren kodieren:
# A tibble: 48 × 5
gen yield block row col
<fct> <dbl> <fct> <dbl> <dbl>
1 st 2972 I 1 1
2 14 2405 I 2 1
3 26 2855 I 3 1
4 ci 2592 I 4 1
5 17 2572 I 5 1
6 wa 2608 I 6 1
7 22 2705 I 7 1
8 13 2391 I 8 1
9 st 3122 II 1 2
10 ci 3023 II 2 2
# ℹ 38 more rowsErkunden
Betrachten wir zunächst die deskriptiven Statistiken. Beachten Sie den Unterschied in der Replikation zwischen Standards und neuen Einträgen:
# A tibble: 33 × 4
gen count mean_yield sd_yield
<fct> <int> <dbl> <dbl>
1 st 6 2759. 832.
2 ci 6 2726. 711.
3 wa 6 2678. 615.
4 19 1 3643 NA
5 11 1 3380 NA
6 07 1 3265 NA
7 03 1 3055 NA
8 04 1 3018 NA
9 01 1 3013 NA
10 30 1 2955 NA
# ℹ 23 more rowsDie drei Standards (ci, st, wa) erscheinen jeweils 6 mal (einmal pro Block), während alle neuen Einträge nur einmal erscheinen. Dies ist das definierende Merkmal eines Augmented Designs.
Schauen wir uns nun die Blockstruktur an:
# A tibble: 6 × 4
block count mean_yield sd_yield
<fct> <int> <dbl> <dbl>
1 VI 8 3205. 417.
2 II 8 2864. 258.
3 IV 8 2797. 445.
4 I 8 2638. 202.
5 III 8 2567. 440.
6 V 8 1390. 207.Wir können Variation zwischen den Blöcken sehen. Block II hat den höchsten mittleren Ertrag, während Block V den niedrigsten hat. Visualisieren wir die Daten mit verschiedenen Farben für Standards und neue Einträge:
# Benutzerdefinierte Farben definieren: Grün für neue Einträge, Rot für Standards
greens30 <- colorRampPalette(c("#bce2cc", "#00923f"))(30)
oranges3 <- colorRampPalette(c("#e4572e", "#ad0000"))(3)
gen_cols <- set_names(c(greens30, oranges3), nm = levels(dat$gen))ggplot(data = dat) +
aes(
y = yield,
x = gen,
color = gen,
shape = block
) +
geom_point() +
scale_x_discrete(
name = "Genotyp"
) +
scale_y_continuous(
name = "Ertrag",
limits = c(0, NA),
expand = expansion(mult = c(0, 0.05))
) +
scale_color_manual(
guide = "none",
values = gen_cols
) +
scale_shape_discrete(
name = "Block"
) +
guides(shape = guide_legend(nrow = 1)) +
theme_classic() +
theme(
legend.position = "top",
axis.text.x = element_text(size = 7)
)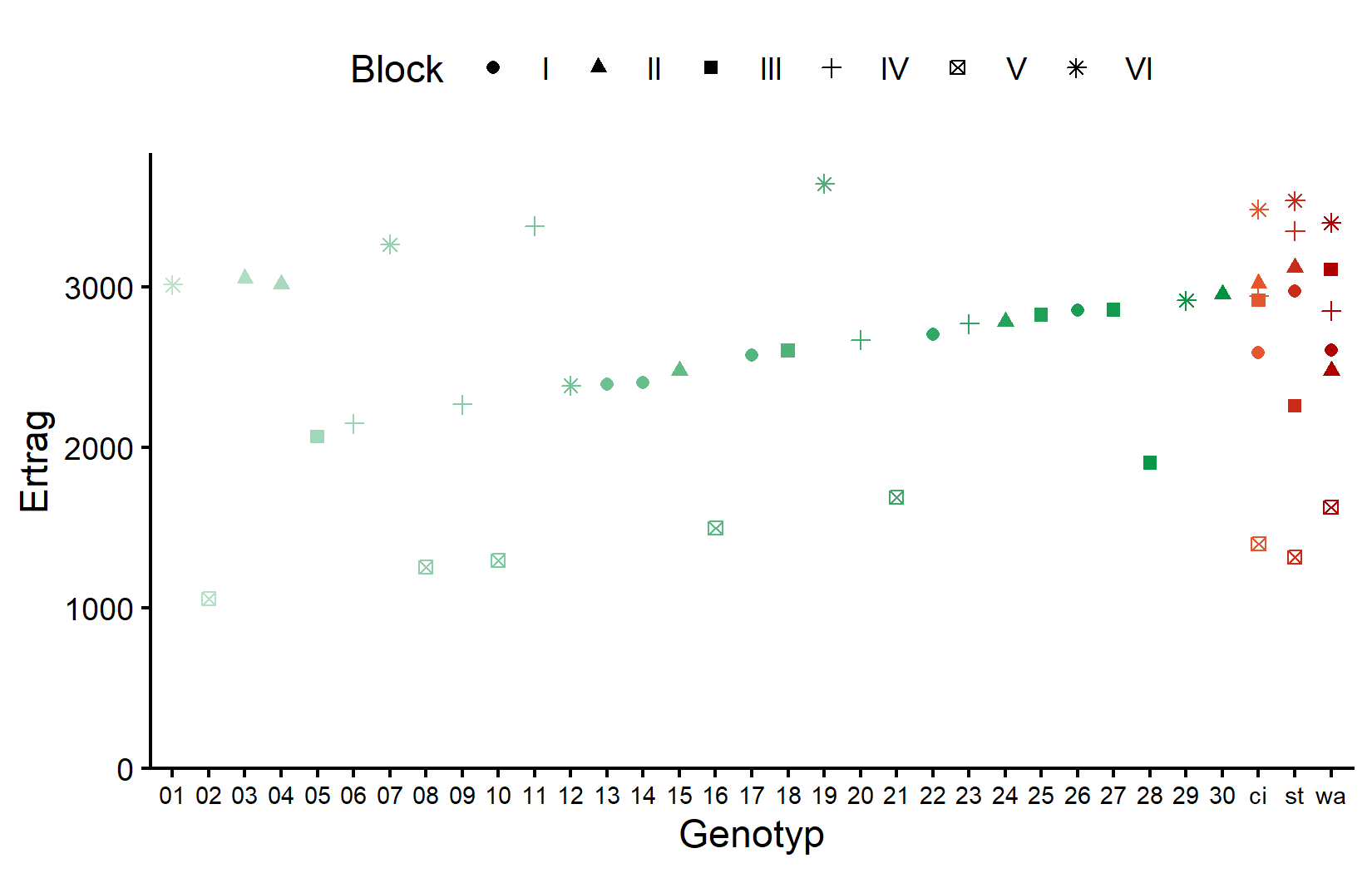
Die Standards (in rot/orange auf der rechten Seite) zeigen Variation über Blöcke hinweg, was uns ermöglicht, Blockeffekte zu schätzen. Schauen wir uns nun das Feldlayout an:
desplot(
data = dat,
flip = TRUE,
form = gen ~ col + row, # Füllfarbe pro Genotyp
col.regions = gen_cols, # benutzerdefinierte Füllfarben
out1 = block, # Linie zwischen Blöcken
text = gen, # Genotypnamen pro Parzelle
cex = 1,
shorten = FALSE,
main = "Feldlayout",
show.key = FALSE
) 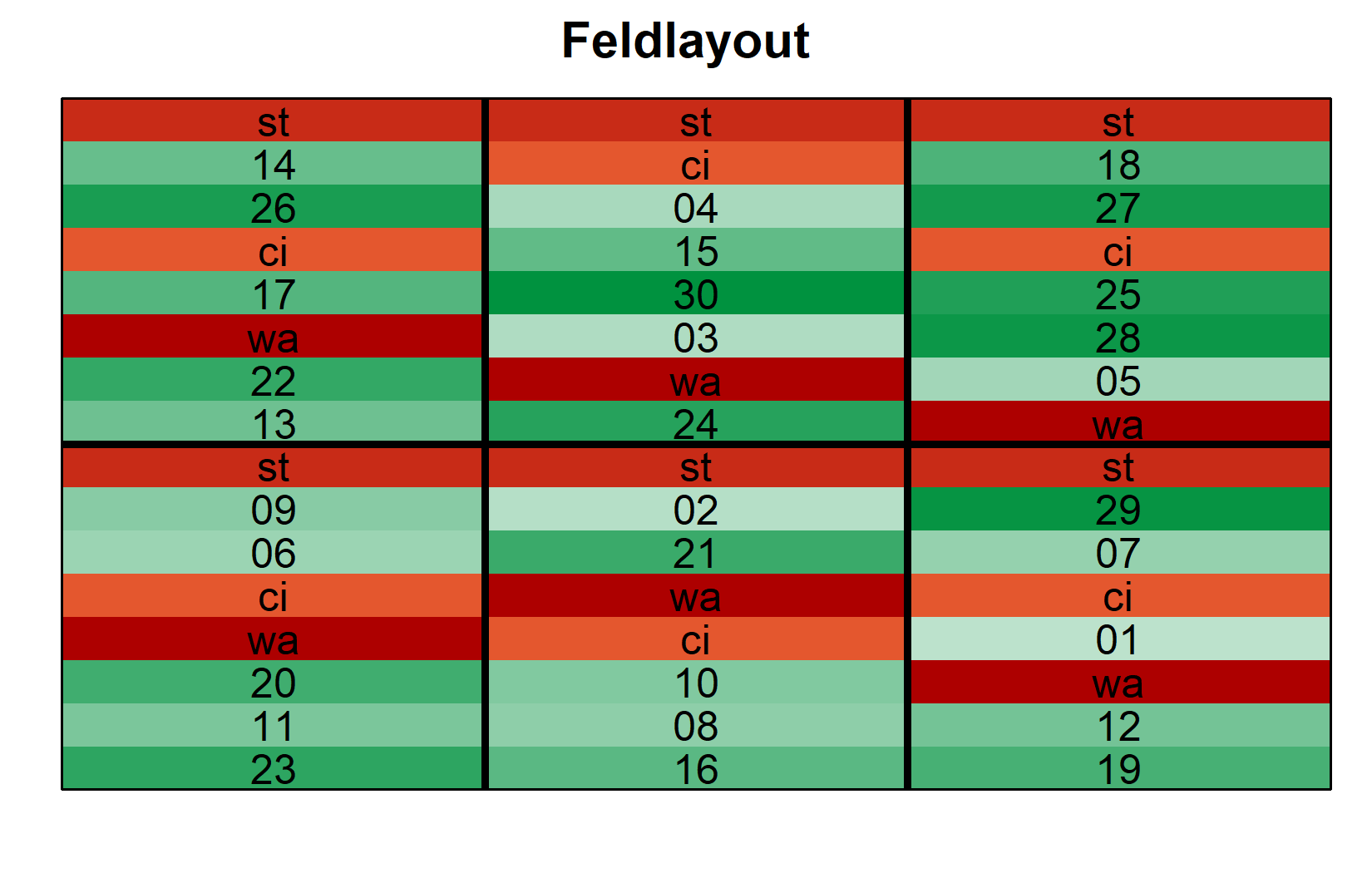
Das Layout zeigt, wie Standards (st, ci, wa) über alle Blöcke verteilt sind, während jeder neue Eintrag nur in einem Block erscheint.
Modell und ANOVA
Modell mit festen Bloecken
Für ein Augmented Design können wir das Modell mit Blöcken als entweder feste oder zufällige Effekte anpassen. Beginnen wir mit festen Blöcken:
mod_fb <- lm(yield ~ gen + block, data = dat)Und vergleichen mit zufälligen Blöcken:
mod_rb <- lmer(yield ~ gen + (1 | block), data = dat)Um zu bestimmen, welches Modell für den Vergleich von Genotypen besser geeignet ist, vergleichen wir den durchschnittlichen Standardfehler einer Differenz (s.e.d.):
# s.e.d. für Modell mit festen Blöcken
sed_fixed <- mod_fb %>%
emmeans(pairwise ~ "gen", adjust = "none") %>%
pluck("contrasts") %>%
as_tibble() %>%
pull("SE") %>%
mean()
# s.e.d. für Modell mit zufälligen Blöcken
sed_random <- mod_rb %>%
emmeans(pairwise ~ "gen", adjust = "none", lmer.df = "kenward-roger") %>%
pluck("contrasts") %>%
as_tibble() %>%
pull("SE") %>%
mean()
tibble(
model = c("Feste Blöcke", "Zufällige Blöcke"),
mean_sed = c(sed_fixed, sed_random)
)# A tibble: 2 × 2
model mean_sed
<chr> <dbl>
1 Feste Blöcke 461.
2 Zufällige Blöcke 462.In diesem Fall hat das Modell mit festen Blöcken einen etwas kleineren s.e.d., daher verwenden wir es für unsere Analyse.
WarnungModellannahmen erfüllt?
An dieser Stelle (d.h. nach dem Modell-Fit und vor der ANOVA-Interpretation) sollte man prüfen, ob die Modellannahmen erfüllt sind. Mehr dazu im Anhang A1: Modelldiagnostik.
Durchführung der ANOVA
ANOVA <- anova(mod_fb)
ANOVAAnalysis of Variance Table
Response: yield
Df Sum Sq Mean Sq F value Pr(>F)
gen 32 12626173 394568 4.331 0.0091056 **
block 5 6968486 1393697 15.298 0.0002082 ***
Residuals 10 911027 91103
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1Der Genotypeffekt ist statistisch signifikant (p < 0.05), was auf Unterschiede zwischen Genotypen hinweist. Der Blockeffekt ist ebenfalls signifikant, was bestätigt, dass die Blockbildung vorteilhaft war.
Mittelwertvergleiche
gen emmean SE df lower.CL upper.CL .group
12 1632 341 10 164 3100 a
06 1823 341 10 355 3291 a
28 1862 341 10 394 3330 a
09 1943 341 10 475 3411 a
05 2024 341 10 556 3492 a
29 2162 341 10 694 3630 a
01 2260 341 10 792 3728 a
15 2324 341 10 856 3792 a
02 2330 341 10 862 3798 a
20 2345 341 10 877 3813 a
13 2388 341 10 920 3856 a
14 2402 341 10 934 3870 a
23 2445 341 10 977 3913 a
07 2512 341 10 1044 3980 a
08 2528 341 10 1060 3996 a
18 2562 341 10 1094 4030 a
10 2568 341 10 1100 4036 a
17 2569 341 10 1101 4037 a
24 2630 341 10 1162 4098 a
wa 2678 123 10 2148 3208 a
22 2702 341 10 1234 4170 a
ci 2726 123 10 2195 3256 a
st 2759 123 10 2229 3289 a
16 2770 341 10 1302 4238 a
25 2784 341 10 1316 4252 a
30 2802 341 10 1334 4270 a
27 2816 341 10 1348 4284 a
26 2852 341 10 1384 4320 a
04 2865 341 10 1397 4333 a
19 2890 341 10 1422 4358 a
03 2902 341 10 1434 4370 a
21 2963 341 10 1495 4431 a
11 3055 341 10 1587 4523 a
Results are averaged over the levels of: block
Confidence level used: 0.95
Conf-level adjustment: sidak method for 33 estimates
P value adjustment: tukey method for comparing a family of 33 estimates
significance level used: alpha = 0.05
NOTE: If two or more means share the same grouping symbol,
then we cannot show them to be different.
But we also did not show them to be the same. Beachten Sie, dass obwohl einige Genotypen höhere adjustierte Mittelwerte haben als andere, mit Tukey-Adjustierung keine signifikanten Unterschiede erkannt werden. Dies liegt teilweise daran, dass nicht replizierte Einträge große Konfidenzintervalle haben. Zum Beispiel hat Genotyp 11 den höchsten adjustierten Mittelwert (3055), aber sein Konfidenzintervall ist breit.
Visualisierung der Ergebnisse
my_caption <- "Punkte repräsentieren Rohdaten (grün = neue Einträge, rot = Standards). Rote Rauten und Fehlerbalken repräsentieren adjustierte Mittelwerte mit 95%-Konfidenzgrenzen pro Genotyp. Mittelwerte, die einen gemeinsamen Buchstaben tragen, unterscheiden sich nicht signifikant nach dem Tukey-Test."
ggplot() +
aes(x = gen) +
# farbige Punkte für die Rohdaten
geom_point(
data = dat,
aes(y = yield, color = gen)
) +
# rote Rauten für die adjustierten Mittelwerte
geom_point(
data = mean_comp,
aes(y = emmean),
shape = 18,
color = "red",
position = position_nudge(x = 0.2)
) +
# rote Fehlerbalken für die Konfidenzgrenzen der adjustierten Mittelwerte
geom_errorbar(
data = mean_comp,
aes(ymin = lower.CL, ymax = upper.CL),
color = "red",
width = 0.1,
position = position_nudge(x = 0.2)
) +
# rote Buchstaben
geom_text(
data = mean_comp,
aes(y = upper.CL, label = str_trim(.group)),
color = "red",
vjust = -0.2,
position = position_nudge(x = 0.2)
) +
scale_color_manual(
guide = "none",
values = gen_cols
) +
scale_x_discrete(
name = "Genotyp",
limits = as.character(mean_comp$gen)
) +
scale_y_continuous(
name = "Ertrag",
limits = c(0, NA),
expand = expansion(mult = c(0, 0.1))
) +
labs(caption = my_caption) +
theme_classic() +
theme(plot.caption = element_textbox_simple(margin = margin(t = 5)),
plot.caption.position = "plot",
axis.text.x = element_text(size = 7))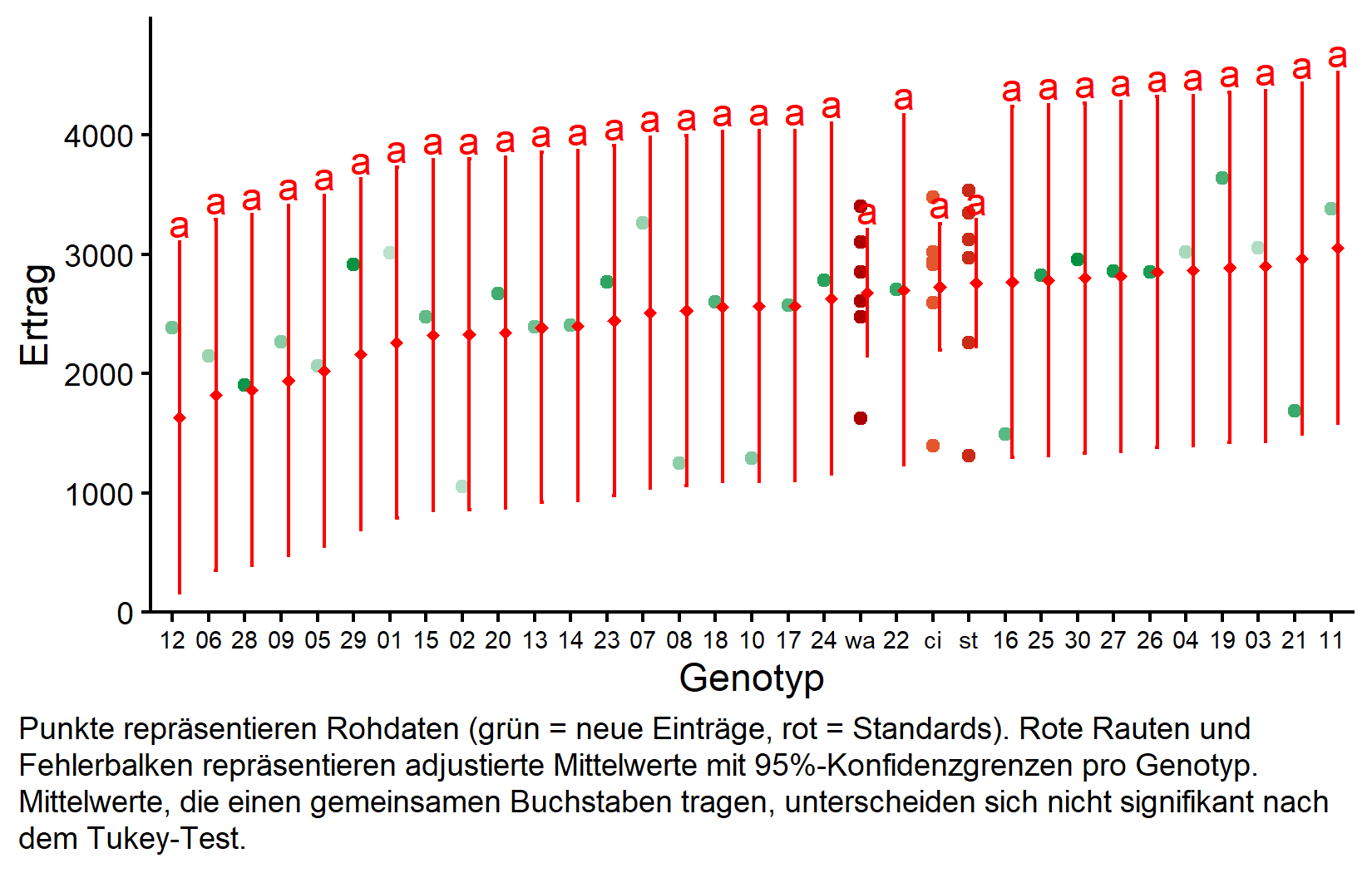
Die Darstellung zeigt deutlich den Unterschied in der Präzision: Standards (auf der rechten Seite) haben viel schmalere Konfidenzintervalle aufgrund der Replikation, während neue Einträge breite Intervalle haben, die auf Einzelbeobachtungen basieren, die für Blockeffekte adjustiert wurden.
Bonus: Varianzkomponenten
Wir können Varianzkomponenten aus beiden Modellen extrahieren, um die Variationsquellen zu verstehen:
# A tibble: 1 × 2
source variance
<chr> <dbl>
1 Residual (festes Modell) 91103.# A tibble: 2 × 2
grp variance
<chr> <dbl>
1 block 434198.
2 Residual 91103.Zusammenfassung
Sie haben nun gelernt, wie man Daten aus einem Augmented Design analysiert, das besonders nützlich ist für das Screening vieler neuer Einträge mit begrenzten Ressourcen.
HinweisWichtige Erkenntnisse
Augmented Designs enthalten replizierte Standards und nicht replizierte neue Einträge, wodurch die Anzahl der Einträge maximiert wird, die getestet werden können.
Standards schätzen Blockeffekte, die dann zur Adjustierung aller Einträge verwendet werden, einschließlich der nicht replizierten.
Der Kompromiss ist die Präzision: Nicht replizierte Einträge haben breitere Konfidenzintervalle als replizierte Standards.
Die Modellwahl (feste vs. zufällige Blöcke) kann darauf basieren, welche den kleineren durchschnittlichen s.e.d. für Genotypvergleiche ergibt.
Praktische Anwendung: Augmented Designs sind ideal für frühe Screening-Versuche, bei denen viele Kandidaten eine erste Bewertung benötigen.
Vorsicht bei der Interpretation: Das Fehlen signifikanter Unterschiede bedeutet nicht, dass Einträge gleich sind - es kann geringe Power für nicht replizierte Vergleiche widerspiegeln.
Literatur
Petersen, Roger G. 1994. Agricultural Field Experiments. CRC Press. https://doi.org/10.1201/9781482277371.
Zitat
Mit BibTeX zitieren:
@online{schmidt2026,
author = {{Dr. Paul Schmidt}},
publisher = {BioMath GmbH},
title = {5. Einfaktorielle ANOVA im Augmented Design},
date = {2026-03-04},
url = {https://biomathcontent.netlify.app/de/content/lin_mod_exp/05_oneway_augmented.html},
langid = {de}
}
Bitte zitieren Sie diese Arbeit als:
Dr. Paul Schmidt. 2026. “5. Einfaktorielle ANOVA im Augmented
Design.” BioMath GmbH. March 4, 2026. https://biomathcontent.netlify.app/de/content/lin_mod_exp/05_oneway_augmented.html.