for (pkg in c("agridat", "desplot", "emmeans", "ggtext", "here", "lme4",
"lmerTest", "multcomp", "multcompView", "tidyverse")) {
if (!require(pkg, character.only = TRUE)) install.packages(pkg)
}
library(agridat)
library(desplot)
library(emmeans)
library(ggtext)
library(here)
library(lme4)
library(lmerTest)
library(multcomp)
library(multcompView)
library(tidyverse)Um alle in diesem Kapitel verwendeten Pakete zu installieren und zu laden, führen Sie folgenden Code aus:
Zeilen-Spalten-Designs
In Kapitel 3 haben wir das Lateinische Quadrat kennengelernt, das zwei Variationsquellen (Zeilen und Spalten) gleichzeitig kontrolliert. Das Lateinische Quadrat hat jedoch eine wesentliche Einschränkung: Die Anzahl der Behandlungen muss gleich der Anzahl der Zeilen und Spalten sein. Dies macht es für Experimente mit vielen Behandlungen unpraktisch.
Was ist ein Zeilen-Spalten-Design?
Ein auflösbares Zeilen-Spalten-Design erweitert das Konzept des Lateinischen Quadrats, um mehr Behandlungen aufzunehmen. Wie ein Alpha-Design hat es vollständige Wiederholungen, die unterteilt sind - aber hier ist jede Wiederholung sowohl in unvollständige Zeilen als auch in unvollständige Spalten unterteilt. Dies bietet eine doppelte Blockbildung innerhalb jeder Wiederholung.
Die wesentlichen Merkmale sind:
- Zweidimensionale Blockbildung: Jede Wiederholung hat sowohl Zeilen- als auch Spaltenstruktur
- Unvollständige Blöcke: Weder Zeilen noch Spalten enthalten alle Behandlungen
- Auflösbarkeit: Wiederholungen sind vollständig und enthalten jede Behandlung genau einmal
- Flexibilität: Kann verschiedene Anzahlen von Behandlungen aufnehmen
Die Vorteile umfassen:
- Kontrolle von zwei Gradienten: Berücksichtigt räumliche Trends in zwei Richtungen gleichzeitig
- Mehr Behandlungen als Lateinisches Quadrat: Nicht auf t×t-Anordnungen beschränkt
- Erhöhte Präzision: Doppelte Blockbildung kann den Versuchsfehler erheblich reduzieren
- Praktisch für Feldversuche: Passt zum rechteckigen Layout vieler Feldexperimente
Daten
Dieses Beispiel betrachtet Daten, die in Kempton, Fox, und Cerezo (1997) veröffentlicht wurden, aus einem Ertragsversuch, der als auflösbares Zeilen-Spalten-Design angelegt wurde. Der Versuch hatte 35 Genotypen (gen), 2 vollständige Wiederholungen (rep) mit jeweils 5 Zeilen (row) und 7 Spalten (col). Somit bildet jede Wiederholung ein 5×7-Raster mit unvollständigen Zeilen und Spalten.
Import
Die Daten sind als Teil des {agridat}-Pakets verfügbar:
dat <- as_tibble(agridat::kempton.rowcol)
dat# A tibble: 68 × 5
rep row col gen yield
<fct> <int> <int> <fct> <dbl>
1 R1 1 1 G20 3.77
2 R1 1 2 G04 3.21
3 R1 1 3 G33 4.55
4 R1 1 4 G28 4.09
5 R1 1 5 G07 5.05
6 R1 1 6 G12 4.19
7 R1 1 7 G30 3.27
8 R1 2 1 G10 3.44
9 R1 2 2 G14 4.3
10 R1 2 4 G21 3.86
# ℹ 58 more rowsDer Datensatz enthält:
-
rep: Zwei vollständige Wiederholungen (R1, R2) -
row: Zeilenposition innerhalb der Wiederholung (1-5) -
col: Spaltenposition innerhalb der Wiederholung (1-7) -
gen: 35 Genotypen -
yield: Ernteertrag
Beachten Sie, dass in diesem Datensatz fehlende Werte vorhanden sind - zwei Parzellen haben keinen erfassten Ertrag.
Formatierung
Für unsere Analyse sollte gen als Faktor kodiert werden. Wir erstellen auch Faktorversionen von row und col für das statistische Modell:
# A tibble: 68 × 7
rep row col gen yield rowF colF
<fct> <int> <int> <fct> <dbl> <fct> <fct>
1 R1 1 1 G20 3.77 1 1
2 R1 1 2 G04 3.21 1 2
3 R1 1 3 G33 4.55 1 3
4 R1 1 4 G28 4.09 1 4
5 R1 1 5 G07 5.05 1 5
6 R1 1 6 G12 4.19 1 6
7 R1 1 7 G30 3.27 1 7
8 R1 2 1 G10 3.44 2 1
9 R1 2 2 G14 4.3 2 2
10 R1 2 4 G21 3.86 2 4
# ℹ 58 more rowsErkunden
Betrachten wir die deskriptiven Statistiken nach Genotyp:
# A tibble: 35 × 4
gen count mean_yield sd_yield
<fct> <int> <dbl> <dbl>
1 G19 2 6.07 1.84
2 G07 2 5.74 0.976
3 G33 2 5.13 0.820
4 G06 2 4.96 0.940
5 G09 2 4.94 1.68
6 G11 2 4.93 1.03
7 G14 2 4.92 0.877
8 G27 2 4.89 1.80
9 G03 2 4.78 0.0424
10 G25 2 4.78 0.361
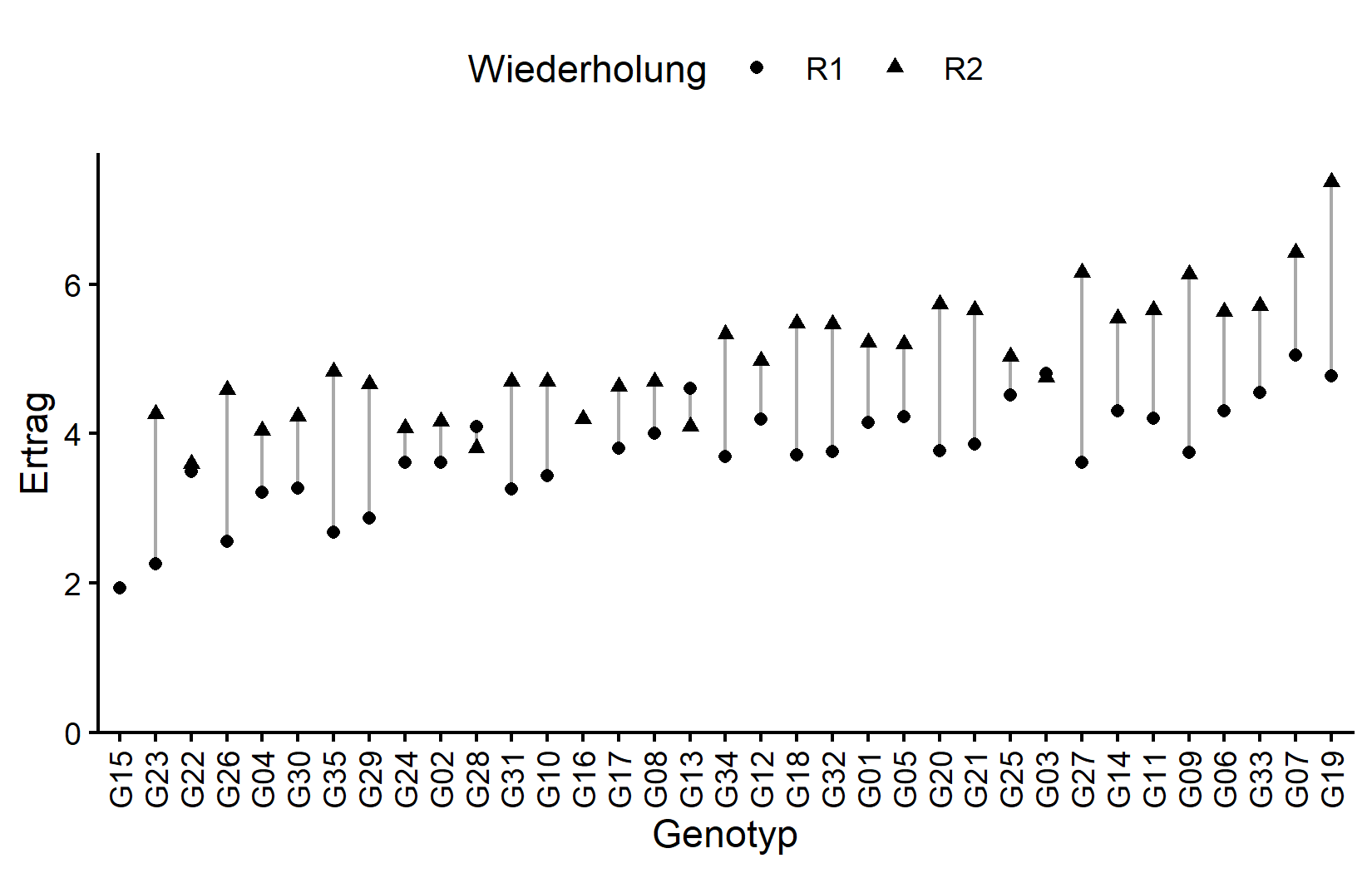
# ℹ 25 more rowsDie meisten Genotypen erscheinen zweimal (einmal pro Wiederholung), aber einige haben aufgrund fehlender Daten nur eine Beobachtung. Visualisieren wir die Daten:
# Genotypen nach mittlerem Ertrag sortieren
gen_order <- dat %>%
group_by(gen) %>%
summarise(mean = mean(yield, na.rm = TRUE)) %>%
arrange(mean) %>%
pull(gen) %>%
as.character()
ggplot(data = dat) +
aes(
y = yield,
x = gen,
shape = rep
) +
geom_line(
aes(group = gen),
color = "darkgrey"
) +
geom_point() +
scale_x_discrete(
name = "Genotyp",
limits = gen_order
) +
scale_y_continuous(
name = "Ertrag",
limits = c(0, NA),
expand = expansion(mult = c(0, 0.05))
) +
scale_shape_discrete(
name = "Wiederholung"
) +
guides(shape = guide_legend(nrow = 1)) +
theme_classic() +
theme(
legend.position = "top",
axis.text.x = element_text(angle = 90, vjust = 0.5)
)
Schauen wir uns nun das Feldlayout an. Beachten Sie, dass die zwei fehlenden Parzellen als weiß/leer erscheinen:
desplot(
data = dat,
form = gen ~ col + row | rep, # Füllfarbe pro Genotyp, Panels pro Wiederholung
text = gen,
cex = 0.7,
shorten = FALSE,
out1 = row, out1.gpar = list(col = "black"), # Linien zwischen Zeilen
out2 = col, out2.gpar = list(col = "black"), # Linien zwischen Spalten
main = "Feldlayout",
show.key = FALSE
) 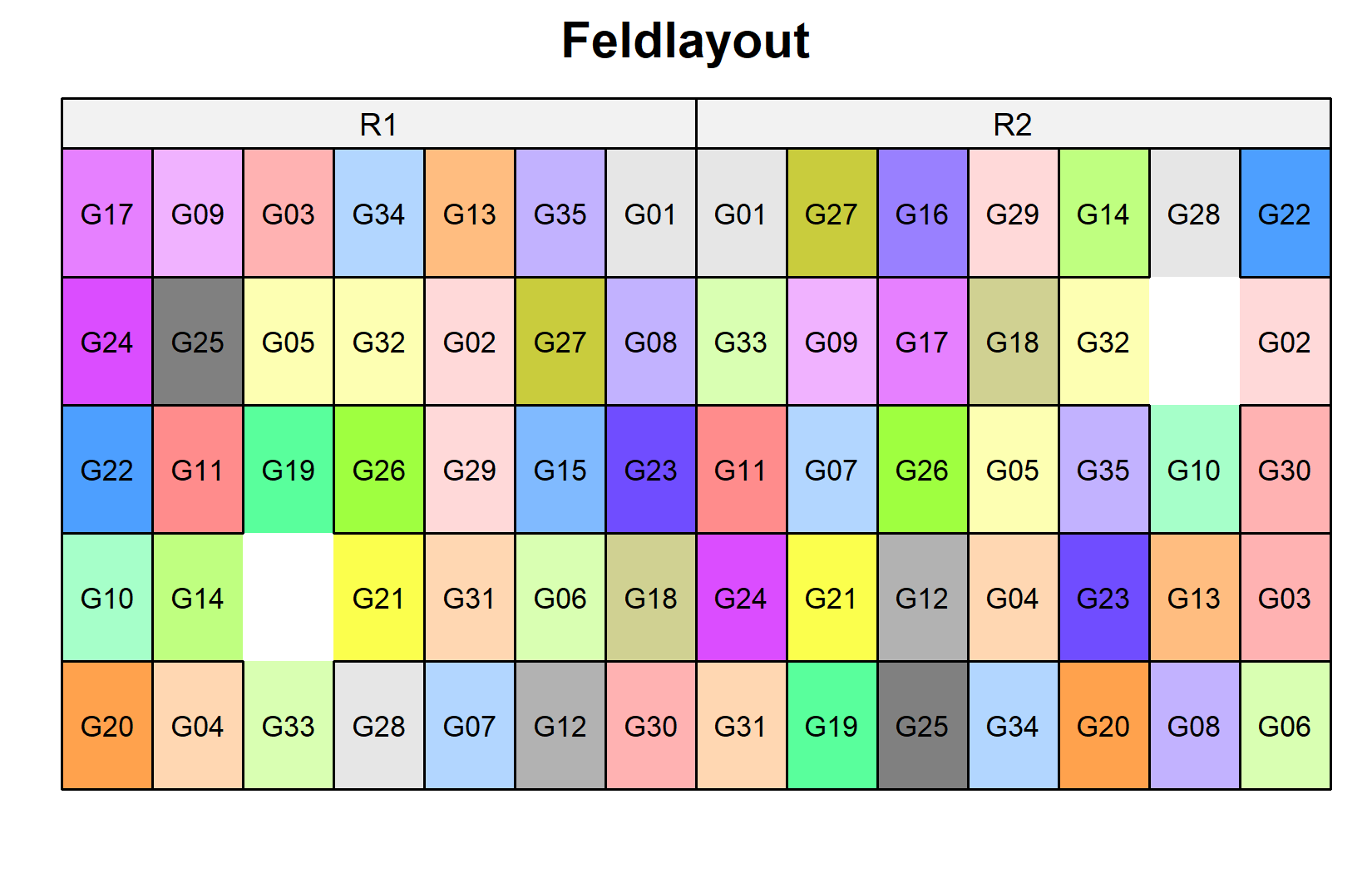
Die schwarzen Linien zeigen die Zeilen- und Spaltenstruktur innerhalb jeder Wiederholung. Jeder Genotyp erscheint einmal pro Wiederholung, aber in verschiedenen Zeilen-Spalten-Positionen.
Modell und ANOVA
Wahl zwischen festen und zufaelligen Effekten
Für ein Zeilen-Spalten-Design muss das Modell Zeilen- und Spalteneffekte innerhalb jeder Wiederholung berücksichtigen. Wir können diese als feste oder zufällige Effekte behandeln. Vergleichen wir beide Ansätze:
Vergleichen wir nun den durchschnittlichen s.e.d. für Genotypvergleiche:
NOTE: A nesting structure was detected in the fitted model:
rowF %in% rep, colF %in% rep# s.e.d. für zufälliges Modell
sed_random <- mod_random %>%
emmeans(pairwise ~ "gen", adjust = "none", lmer.df = "kenward-roger") %>%
pluck("contrasts") %>%
as_tibble() %>%
pull("SE") %>%
mean()
tibble(
model = c("Feste Zeilen/Spalten", "Zufällige Zeilen/Spalten"),
mean_sed = c(sed_fixed, sed_random)
)# A tibble: 2 × 2
model mean_sed
<chr> <dbl>
1 Feste Zeilen/Spalten 0.408
2 Zufällige Zeilen/Spalten 0.402In diesem Fall hat das Modell mit festen Effekten einen etwas kleineren s.e.d., daher verwenden wir es für unsere Analyse.
WarnungModellannahmen erfüllt?
An dieser Stelle (d.h. nach dem Modell-Fit und vor der ANOVA-Interpretation) sollte man prüfen, ob die Modellannahmen erfüllt sind. Mehr dazu im Anhang A1: Modelldiagnostik.
Durchführung der ANOVA
ANOVA <- anova(mod_fixed)
ANOVAAnalysis of Variance Table
Response: yield
Df Sum Sq Mean Sq F value Pr(>F)
gen 34 32.157 0.9458 10.7456 4.969e-05 ***
rep 1 24.901 24.9014 282.9193 1.042e-09 ***
rep:rowF 8 2.512 0.3140 3.5680 0.023647 *
rep:colF 12 6.327 0.5273 5.9905 0.002067 **
Residuals 12 1.056 0.0880
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1Der Genotypeffekt ist nicht statistisch signifikant (p > 0.05), was auf keine starke Evidenz für Unterschiede zwischen Genotypen in diesem Versuch hinweist. Dennoch untersuchen wir die Mittelwertvergleiche.
Mittelwertvergleiche
gen emmean SE df lower.CL upper.CL .group
G04 3.48 0.270 12 2.89 4.07 a
G23 3.58 0.270 12 2.99 4.17 ab
G15 3.60 0.442 12 2.64 4.57 abcde
G35 3.63 0.277 12 3.03 4.24 abc
G31 3.79 0.280 12 3.18 4.40 abcd
G02 3.84 0.279 12 3.23 4.45 abcd
G26 3.90 0.291 12 3.26 4.53 abcde
G24 3.90 0.267 12 3.32 4.49 abcd f
G29 3.91 0.276 12 3.31 4.51 abcde
G30 3.99 0.270 12 3.40 4.58 abcdefg
G32 4.12 0.276 12 3.52 4.72 abcdefgh
G17 4.14 0.282 12 3.53 4.75 abcdefgh
G09 4.15 0.274 12 3.56 4.75 abcdefgh
G34 4.20 0.267 12 3.62 4.78 abcdefgh
G16 4.23 0.432 12 3.29 5.17 abcdefghij
G05 4.25 0.278 12 3.64 4.85 abcdefghi
G20 4.25 0.266 12 3.67 4.83 abcdefgh
G22 4.27 0.282 12 3.66 4.88 abcdefghi
G10 4.36 0.278 12 3.76 4.97 abcdefghij
G28 4.37 0.278 12 3.77 4.98 bcdefghij
G18 4.48 0.284 12 3.86 5.10 cdefghij
G21 4.57 0.269 12 3.98 5.16 defghij
G08 4.58 0.285 12 3.95 5.20 defghij
G25 4.59 0.277 12 3.98 5.19 defghij
G13 4.73 0.284 12 4.11 5.35 e ghijkl
G27 4.75 0.282 12 4.13 5.36 fghijk
G33 4.76 0.286 12 4.13 5.38 e ghijk
G14 4.79 0.270 12 4.20 5.38 ghijkl
G01 4.88 0.268 12 4.30 5.46 hijkl
G07 4.94 0.270 12 4.35 5.53 hijkl
G11 4.97 0.276 12 4.37 5.57 hijkl
G12 5.13 0.293 12 4.49 5.77 ijkl
G03 5.15 0.281 12 4.54 5.76 jkl
G06 5.53 0.280 12 4.92 6.14 kl
G19 5.60 0.281 12 4.99 6.22 l
Results are averaged over the levels of: colF, rowF, rep
Confidence level used: 0.95
significance level used: alpha = 0.05
NOTE: If two or more means share the same grouping symbol,
then we cannot show them to be different.
But we also did not show them to be the same. Die Kompaktbuchstabendarstellung zeigt die Gruppierungen basierend auf Fishers LSD-Test. Bei 35 Genotypen wird die Buchstabendarstellung komplex, bietet aber dennoch eine prägnante Zusammenfassung, welche Genotypen sich signifikant unterscheiden.
Visualisierung der Ergebnisse
my_caption <- "Schwarze Punkte repräsentieren Rohdaten. Rote Rauten und Fehlerbalken repräsentieren adjustierte Mittelwerte mit 95%-Konfidenzgrenzen pro Genotyp. Mittelwerte, die einen gemeinsamen Buchstaben tragen, unterscheiden sich nicht signifikant nach Fishers LSD-Test."
ggplot() +
aes(x = gen) +
# schwarze Punkte für die Rohdaten
geom_point(
data = dat,
aes(y = yield)
) +
# rote Rauten für die adjustierten Mittelwerte
geom_point(
data = mean_comp,
aes(y = emmean),
shape = 18,
color = "red",
position = position_nudge(x = 0.2)
) +
# rote Fehlerbalken für die Konfidenzgrenzen der adjustierten Mittelwerte
geom_errorbar(
data = mean_comp,
aes(ymin = lower.CL, ymax = upper.CL),
color = "red",
width = 0.1,
position = position_nudge(x = 0.2)
) +
# rote Buchstaben
geom_text(
data = mean_comp,
aes(y = upper.CL, label = str_trim(.group)),
color = "red",
angle = 90,
hjust = -0.2,
position = position_nudge(x = 0.2)
) +
scale_x_discrete(
name = "Genotyp",
limits = as.character(mean_comp$gen)
) +
scale_y_continuous(
name = "Ertrag",
expand = expansion(mult = c(0, 0.05))
) +
coord_cartesian(ylim = c(0, NA)) +
labs(caption = my_caption) +
theme_classic() +
theme(plot.caption = element_textbox_simple(margin = margin(t = 5)),
plot.caption.position = "plot",
axis.text.x = element_text(angle = 90, vjust = 0.5))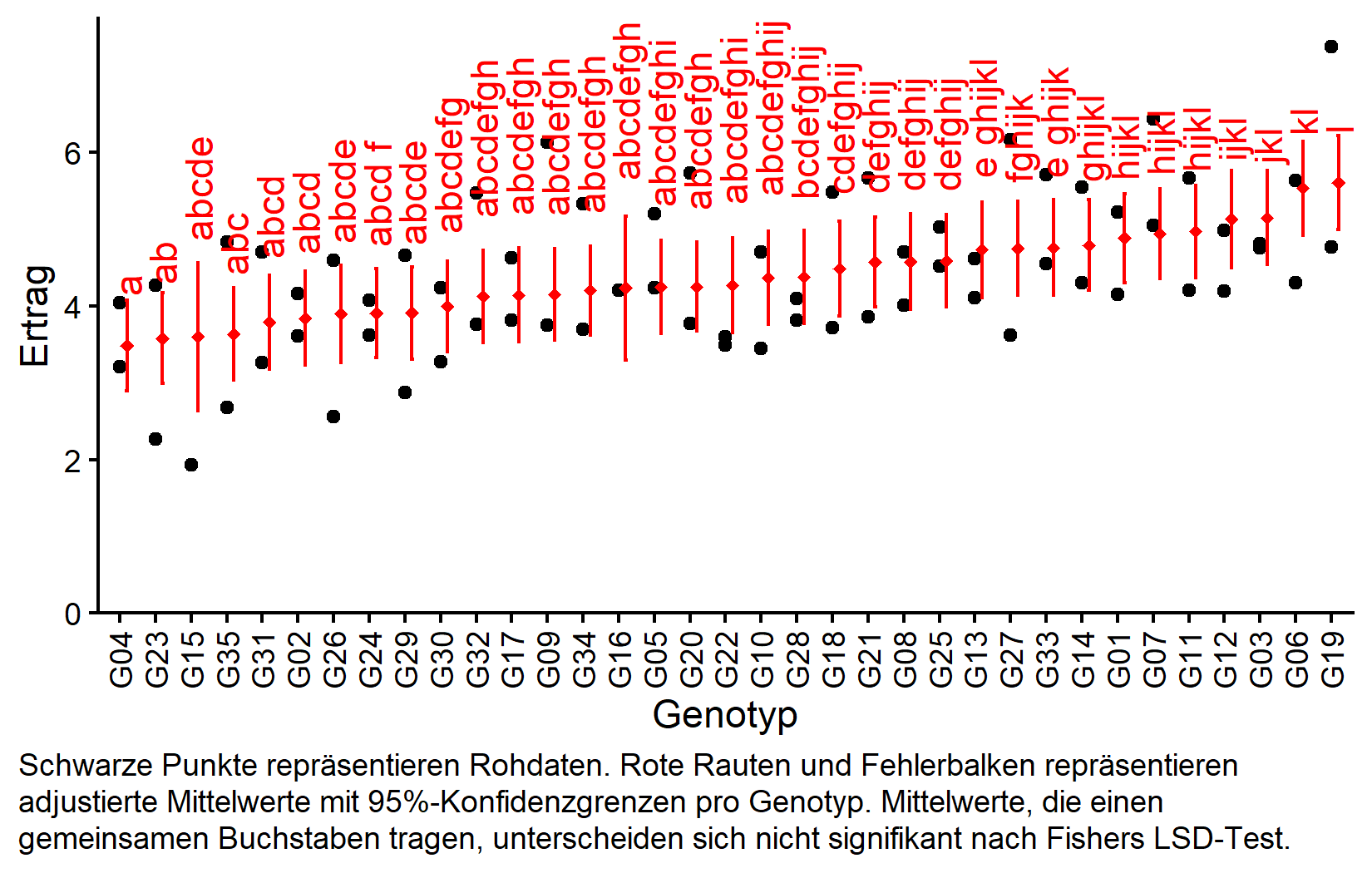
Bonus: Designeffizienz
Bewerten wir die Effizienz des Zeilen-Spalten-Designs im Vergleich zu einem einfachen RCBD (unter Ignorierung der Zeilen- und Spaltenstruktur):
[1] 1.953932Eine Effizienz > 1 zeigt an, dass das Zeilen-Spalten-Design effizienter ist als ein einfaches RCBD, was bedeutet, dass die Zeilen- und Spaltenblockierung den Versuchsfehler erfolgreich reduziert hat.
Zusammenfassung
Sie haben nun gelernt, wie man Daten aus einem auflösbaren Zeilen-Spalten-Design analysiert, das eine leistungsstarke Kontrolle über zwei Quellen räumlicher Variation bietet.
HinweisWichtige Erkenntnisse
Zeilen-Spalten-Designs kontrollieren zwei Variationsquellen gleichzeitig durch Blockbildung sowohl in Zeilen- als auch in Spaltenrichtung innerhalb jeder Wiederholung.
Flexibler als das Lateinische Quadrat: Kann jede Anzahl von Behandlungen aufnehmen, nicht auf t×t-Anordnungen beschränkt.
Doppelte Blockbildung innerhalb von Wiederholungen bietet erhöhte Präzision, wenn räumliche Trends in zwei Richtungen existieren.
Die Modellwahl zwischen festen und zufälligen Zeilen-/Spalteneffekten kann darauf basieren, welche den kleineren durchschnittlichen s.e.d. ergibt.
Das Modell mit festen Effekten:
yield ~ gen + rep + rep:rowF + rep:colF(Zeilen und Spalten genestelt innerhalb von Wiederholungen).Eine Designeffizienz > 1 im Vergleich zum RCBD bestätigt den Vorteil der zusätzlichen Zeilen-Spalten-Struktur.
Umgang mit fehlenden Daten: Zeilen-Spalten-Designs können auch bei fehlenden Beobachtungen analysiert werden, obwohl die Präzision beeinträchtigt sein kann.
Zusammenfassung des Designvergleichs
Fassen wir die Progression der in dieser Kapitelserie behandelten Designs zusammen:
| Design | Blockstruktur | Modellformel | Geeignet für |
|---|---|---|---|
| CRD | Keine | y ~ trt |
Homogene Bedingungen |
| RCBD | Vollständige Blöcke | y ~ trt + block |
Ein Gradient |
| Lateinisches Quadrat | Zeilen + Spalten (vollständig) | y ~ trt + row + col |
Zwei Gradienten, wenige Behandlungen |
| Alpha-Design | Unvollständige Blöcke in Wdh. | y ~ trt + rep + (1|rep:block) |
Viele Behandlungen, ein Gradient |
| Augmented | Standards + Einträge | y ~ trt + block |
Screening vieler nicht replizierter Einträge |
| Zeilen-Spalten | Unv. Zeilen + Spalten in Wdh. | y ~ trt + rep + rep:row + rep:col |
Viele Behandlungen, zwei Gradienten |
Literatur
Kempton, R. A., P. N. Fox, und M. Cerezo. 1997. Statistical Methods for Plant Variety Evaluation. Springer Netherlands. https://doi.org/10.1007/978-94-009-1503-9.
Zitat
Mit BibTeX zitieren:
@online{schmidt2026,
author = {{Dr. Paul Schmidt}},
publisher = {BioMath GmbH},
title = {6. Einfaktorielle ANOVA im Zeilen-Spalten-Design},
date = {2026-03-04},
url = {https://biomathcontent.netlify.app/de/content/lin_mod_exp/06_oneway_rowcol.html},
langid = {de}
}
Bitte zitieren Sie diese Arbeit als:
Dr. Paul Schmidt. 2026. “6. Einfaktorielle ANOVA im
Zeilen-Spalten-Design.” BioMath GmbH. March 4, 2026. https://biomathcontent.netlify.app/de/content/lin_mod_exp/06_oneway_rowcol.html.